Benutzer:Bin im Garten/Chemie/Brückenkurs
Analytische Chemie Bearbeiten
Die Analytische Chemie beschäftigt sich als Teilgebiet der Chemie mit der Identifizierung und der Mengenbestimmung von chemischen Substanzen (in diesem Zusammenhang als Analyten bezeichnet). Sie spielt in fast allen chemischen Teildisziplinen eine bedeutende Rolle und ist häufig Gegenstand aktueller öffentlicher Diskussionen wie zum Beispiel in der Umweltanalytik.
Grundlegende Typen der Analytischen Chemie Bearbeiten
Die wohl wichtigste Unterscheidung ist die zwischen qualitativer Analyse, quantitativer Analyse und Strukturanalytik:
- Die qualitative Analyse fragt nach dem Was im Sinne von „Was für ein Stoff ist das?“ Liegt nicht nur eine chemische Verbindung vor, sondern ein Gemisch, lautet die Frage „Welche Substanzen sind in der Probe vorhanden?“. Grundaufgabe der qualitativen Analyse ist also die Identifikation von Stoffen (Durchführung einer Nachweisreaktion, ggf. nach vorheriger Entstörung oder Auftrennung).
- Die quantitative Analyse fragt dagegen nach dem Wie viel, d. h. danach, welche Menge eines Stoffes (des Analyten) in einem Gemisch (der Probe) vorhanden ist.
Was „wie viel“ genau bedeuten soll, ist übrigens gar nicht so trivial. Meist ist hier die Stoffmengenkonzentration gemeint, also zum Beispiel die Angabe, wie viel mol Coffein pro Liter Kaffee vorliegen. - Die Strukturanalyse fragt nach dem molekularen Aufbau einer Substanz (der chemischen Strukturformel oder der Kristallstruktur)
Qualitative und quantitative Analytik werden oft aufeinander aufbauend durchgeführt: Für die quantitative Analyse sollte die zu bestimmende Substanz idealerweise bekannt sein. Voraussetzung für eine qualitative Analyse ist eine genügend große Menge Analyt in der Probe, abhängig von der Nachweisgrenze der verwendeten Methode. Eine Sonderstellung nimmt die Strukturbestimmung ein. Mit dem Aufkommen moderner Kopplungsmethoden (s.u.) werden aber Struktur-bestimmende Analyseverfahren auch in der qualitativen und quantitativen Analytik immer wichtiger.
Neben der Bestimmung einzelner Stoffe eines Gemischs werden oftmals Summenparameter bestimmt – insbesondere wenn es um schnelle Grundaussagen über eine Probe geht. Beispiele sind der TOC (Total Organic Carbon, ein Maß für den Gesamtgehalt organischer Verbindungen), der CSB (Chemischer Sauerstoffbedarf als Maß für die Gesamtmenge an oxidierbaren Substanzen)oder der TEAC-Assay (antioxidative Kapazität einer Probe).
In der Polymeranalytik ist speziell die Molekulargewichtsverteilung der Polymere von Interesse, da Polymere niemals aus Molekülen gleicher Molekülmasse bestehen, sondern um einen statistischen Mittelwert verteilt sind; diese mittlere Molekülgröße beziehungsweise die Molekulargewichtsverteilung sind hier spezifische Eigenschaften des Polymers.
Schließlich gibt es noch die verschiedenen Verfahren der Oberflächenanalytik. Das besondere an diesen analytischen Methoden ist, dass sie besonders sensitiv und zugleich selektiv Oberflächen-Eigenschaften abbilden können. Beispiele für diese Methoden sind: Elektronen-Energieverlustspektroskopie (EELS), Röntgen-Photoelektronen-Spektroskopie (XPS), Auger-Elektronen-Spektroskopie (AES), Ultraviolet-Photoelektronen-Spektroskopie (UPS), Niederenergetische Ionenstreuspektroskopie (ISS=LEIS), Rutherford Backscattering Spectrometry (RBS), (Surface) Extended X-Ray absorption Fine Structure ((S)EXAFS=XANES), Röntgen-Nahkanten-Absorptions-Spektroskopie (XANES=NEXAFS) oder Beugung niederenergetischer Elektronen (LEED).
Nass-chemische Analysemethoden Bearbeiten
Die nass-chemische Analytik bedient sich bei der Identifikation und Quantifizierung ausschließlich chemischer Methoden; irgendwelche Instrumente, die physikalische Methoden zu Hilfe nehmen, werden nicht benutzt, was aber Geräte zur Automatisierung der Analysen (zum Beispiel Continuous Flow Analysis) nicht ausschließt. Beispiele für qualitative Methoden sind:
- Nachweisreaktionen
farbige Komplexbildungsreaktionen oder Niederschlägen durch Fällungsreaktionen - Flammenfärbung
Beispiel: viele Metallionen färben eine Bunsenbrennerflamme in charakteristischer Weise
Aber auch quantitative Bestimmungen lassen sich rein chemisch durchführen:
- Photometrie
Der Analyt reagiert mit einem Reaktionspartner unter Bildung eines farbigen Komplexes. Die Stärke der Färbung wird anschließend mit der Färbung von Lösungen bekannter Konzentration verglichen. - Titration (Volumetrie)
Zu einer Lösung des Analyten wird die Lösung eines Reaktionspartners bekannter Konzentration langsam zugegeben. Wenn der Analyt vollständig abreagiert ist, bewirkt der zugesetzte Reaktionspartner bzw. ein Indikator einen Farbumschlag, eine Niederschlagsbildung oder sonst ein deutlich sichtbares Ereignis. Aus dem Volumen der verbrauchten Lösung des Reaktionspartners kann man die Konzentration des Analyten errechnen. - Gravimetrie
Der Analyt reagiert mit einem Reaktionspartner und bildet einen unlöslichen Niederschlag bekannter Zusammensetzung; aus dessen Gewicht wird die Analytmenge bestimmt (daher der Name: gravis ist Latein und bedeutet „schwer“).
Instrumentelle Analytik Bearbeiten
→ Hauptartikel: Instrumentelle Analytik
Wichtiger als die rein chemischen Nachweise sind heutzutage die Methoden der instrumentellen chemischen Analytik, deren Anzahl fast schon unüberschaubar geworden ist. Die Verfahren beruhen im Wesentlichen auf physikalischen Messprinzipien. Viele dieser Methoden sind sowohl für qualitative als auch quantitative Bestimmungen verwendbar. Auch hier nur ein paar Beispiele:
- Spektroskopie
Hier wird die Wellenlängen-abhängige Absorption oder Emission von elektromagnetischer Strahlung benutzt, die für den jeweiligen Analyten charakteristisch ist. Elektromagnetische Strahlung kann dabei sichtbares oder UV-Licht sein (UV/VIS-Spektroskopie), infrarotes Licht (IR-Spektroskopie, Raman-Spektroskopie), Röntgenstrahlung (Röntgen-Fluoreszenz Analyse, RFA) oder Gamma-Strahlung (Mößbauer-Effekt). Zur quantitativen Elementanalytik kommen hauptsächlich zum Einsatz Atomabsorptionsspektroskopie, Atomemissionsspektroskopie und induktiv gekoppelte Plasmen gekoppelt mit Optischer Emissionsspektroskopie (ICP-OES) oder gekoppelt mit Massenspektrometrie (ICP-MS) . - Massenspektrometrie (MS)
Die Analyt-Moleküle werden auf irgendeine Weise ionisiert und im Vakuum in die Gasphase gebracht. Die Massen der intakten Molekülionen und der sog. Fragmentionen (Molekülionen können zerbrechen und dabei Fragmente bilden) werden bestimmt. Es gibt eine Unmenge an verschiedenen Ionisierungsmethoden und Detektortypen (Elektronenstoß-Ionisation (EI), chemische Ionisation (CI), Elektrospray (ESI), Atmospheric Pressure Chemical Ionization (APCI), Matrix Assisted Laser Desorption Ionisation (MALDI), Fast Atom Bombardment (FAB), Sekundärionen-Massenspektrometrie (SIMS); Felddesorption (FD), Feldionisation (FI), Thermische Ionisation (TIMS), ICP-Massenspektrometrie (ICP-MS); Sektorfeld-Massenspektrometer, Quadrupol-Massenspektrometer, Flugzeit-Massenspektrometer, Ionenfallen-Massenspektrometer etc.). - Kernresonanz-Spektroskopie (NMR)
Bei dieser besonderen Art der Spektroskopie werden magnetische Wechselwirkungen zwischen Atomkernen und Elektronen in den Analyt-Molekülen ausgenutzt. Es gibt eine unüberschaubare Zahl an speziellen Detektionsmethoden (zum Beispiel COESY, NOESY), sog. 1D-, 2D- und 3D-NMR etc. Eine besondere Spielart der NMR ist die sog. MRT (Magnet-Resonanz-Tomographie), die als bildgebendes Verfahren in der Medizin erhebliche Bedeutung gewonnen hat. - Chromatographie
Ziel ist hier die Trennung verschiedener Substanzen. Dazu wird das Analytgemisch in einem Lösungsmittel (mobile Phase) gelöst, das dann eine feste Trägersubstanz (stationäre Phase) durchströmt (Flüssigchromatographie). Alternativ kann das Analytgemisch auch verdampft an der stationären Phase vorbei geführt werden (Gaschromatographie). Durch unterschiedlich starke Wechselwirkungen mit der stationären Phase werden manche Analyten schnell, andere langsam in Flussrichtung transportiert. Die Wanderungsgeschwindigkeit ist für den jeweiligen Analyten charakteristisch. - Elektroanalytische Messmethoden
Hier werden elektrochemische Parameter (Redoxpotentiale, elektrischer Strom, Leitfähigkeit etc.) benutzt, um qualitative und quantitative Analysen durchzuführen. Stichworte sind Voltammetrie/Polarographie, Coulometrie, Amperometrie, Potentiometrie, Konduktometrie, Elektrogravimetrie etc.
Spektroskopische Methoden haben über ihre Anwendung in der klassischen Analytik hinaus erhebliche Bedeutung für die Strukturaufklärung chemischer Verbindungen. Insbesondere die Kombination mehrerer spektroskopischer Methoden ist vor allem in der Organischen Chemie ein sehr effektives Werkzeug. Daneben spielt die Röntgenstrukturanalyse eine bedeutende Rolle bei der Aufklärung von Kristallstrukturen.
In der Praxis finden sich sehr oft Überschneidungen von nass-chemischer und instrumenteller Analytik: Häufig wird eine Probe zunächst nass-chemisch aufbereitet, um für eine instrumentelle Methode verwendbar zu sein. So müssen schwer verdampfbare Substanzen chemisch modifiziert werden (Derivatisierung), damit sie mittels Gaschromatographie analysiert werden können, oder es müssen besonders komplexe Gemische zunächst mit nass-chemischen Methoden aufgetrennt werden, bevor die instrumentelle Analytik zum Zuge kommen kann.
Anwendungen Bearbeiten
Die vielen verschiedenen Analysemethoden erlauben eine Vielzahl von Anwendungen, beispielsweise:
- Die Strukturaufklärung dient der Identifizierung neuer chemischer Verbindungen bei der chemischen Synthese oder bei der Erforschung neuer Naturstoffe und dem Verständnis ihrer Eigenschaften.
- Besonders in der Umwelt- und Lebensmittelanalytik wurden in den letzten Jahren enorme Fortschritte in der Leistungsfähigkeit analytischer Messmethoden und deren Nachweisgrenzen gemacht. Hier, sowie in der forensischen Chemie müssen Substanzen identifiziert und quantifiziert werden.
- Bei der Herstellung chemischer, pharmazeutischer und kosmetischer Produkte sowie von Nahrungsmitteln sind im Rahmen der Qualitätskontrolle chemische Analysen unumgänglich.
Literatur Bearbeiten
- Jander, Blasius, Strähle, Schweda: Lehrbuch der analytischen und präparativen anorganischen Chemie; Hirzel, Stuttgart; Auflage: 16., überarb. A. (März 2006); ISBN 978-3-7776-1388-8
- Jander, Blasius, Strähle: Einführung in das anorganisch-chemische Praktikum (einschl. der quantitativen Analyse); Hirzel, Stuttgart; Auflage: 15., neu bearb. Aufl. (Oktober 2005); ISBN 978-3-7776-1364-2
- Otto: Analytische Chemie; Wiley-VCH; Auflage: 3., vollst. überarb. u. erw. A. (Juli 2006); ISBN 978-3-527-31416-4
- Skoog, Leary: Instrumentelle Analytik. Grundlagen, Geräte, Anwendungen (Springer-Lehrbuch); Springer, Berlin; Auflage: 1 (Mai 1996); ISBN 978-3-540-60450-1
- Schwedt: Analytische Chemie; Wiley-VCH; Auflage: 1 (2004); ISBN 978-3-527-30866-8
- Handbuch der experimentellen Chemie Sekundarbereich II, Band 3 + 4, Analytische Chemie und Umweltanalytik I + II, Aulis Verlag Deubner & Co. KG * Köln
Weblinks Bearbeiten
- Linkkatalog zum Thema Bin im Garten/Chemie/Brückenkurs bei curlie.org (ehemals DMOZ)
- Fachgruppe Analytische Chemie der GDCh
- aktuelle Wochenschau - Neuigkeiten aus der analytischen Forschung
- Verzeichnis frei zugänglicher Lehrangebote in Analytischer Chemie
Qualitative Analyse Bearbeiten
Die qualitative Analyse beschäftigt sich mit dem Nachweis chemischer Elemente, funktioneller Gruppen oder Verbindungen, ohne deren Mengenverhältnisse zu berücksichtigen. Dieser geschieht durch Nachweisreaktionen oder auf instrumentellem Wege.
Anfänge, Entwicklung und Methoden der Analytischen Chemie Bearbeiten
Immer wieder stießen Menschen auf unbekannte Flüssigkeiten, Gegenstände, Nahrungsmittel und Getränke, deren unbekannte Wirkung sie untersuchen wollten. Während einige vorsichtige Regierende in Antike und Mittelalter zu derlei lebensmittelanalytischen Zwecken Sklaven als Vorkoster einsetzten - und wohl auch verbrauchten - , hielten sich andere Monarchen an ihren Höfen oft Gelehrte. Probier- und Experimentierkünstler, Hofastrologen und -theologen, Ärzte, Kräuterfrauen, Quacksalber, Alchemisten, Meister und Magier gaben ihre oft geheimen Entdeckungen und Erfahrungen von Generation zu Generation weiter - und mit dem Aufkommen naturwissenschaftlicher Untersuchungs- und Forschungsmethoden entwickelten sich erste systematische Vorgehensweisen zur Untersuchung unbekannter Proben auf die in ihnen enthaltenen Stoffe.

Eine der wohl ältesten physikalisch-analytischen Methoden zur Untersuchung einer unbekannten Metallprobe war vielleicht das archimedische Prinzip: der Vergleich der Dichte durch Untertauchen in Wasser, um echtes von verfälschtem Gold unterscheiden zu können.
Auch chemische Analysemethoden gab es schon, noch bevor sich die Chemie als Naturwissenschaft etablierte. Schon Plinius wusste Eisensulfat in Grünspan nachzuweisen, indem er Galläpfelsaft einsetzte (dieser bildet mit Eisen-II-Ionen eine schwarze Eisenverbindung).
Nach Entdeckung der Salpeter- und Schwefelsäure in den Vitriol-, Alaun- und Salpetersiedereien (Byzanz, 13. Jahrhundert) konnte man Silber- und Goldlegierungen auch chemisch voneinander unterscheiden: Silber löst sich in Salpetersäure („Scheidewasser“) auf, Gold nicht. Als man dann Salmiak in Salpetersäure löste, war das Königswasser entdeckt (Venedig, 15. Jahrhundert) - es löste selbst den König der Metalle auf, das Gold. Und mit Hilfe von Kupfersalzlösungen - hergestellt z.B. aus Scheidewasser und Bronze - lehrte Andreas Libavius (ca. 1550-1616), wie man Salmiakgeist im Wasser nachweist: Kupfersalzlösungen färben sich durch den Salmiakgeist tiefblau.
Robert Boyle entwickelte 1685 den folgenden ersten Analysengang zur Untersuchung der Qualität eines Gewässers ohne gesundheitsschädliche Geschmacksproben:
- Messung der Temperatur und Bestimmung der Dichte (Volumen abmessen und Wiegen)
- Vorsichtige Bestimmung von Farbe, Geruch und Wirkung auf die Haut
- Ermittlung beweglicher Teilchen im Wasser mit Hilfe von Lupen und der Wirkung von Luft auf die Wasserprobe,
- Test mit Galläpfelsaft (sind im Wasser Eisensalze enthalten, so färbt es sich schwarz, mit Kupfersalzen auch rot und/oder trüb),
- Test mit Veilchen- oder Rotkohlsaft (Anthocyane als Indikator)

Friedrich Hoffmann erweiterte den Analysengang 1703 um den Nachweis von Kochsalz (Nachweismittel: Höllenstein) und Schwefelverbindungen (mit Hilfe von Quecksilber und/oder Quecksilbersalzen), und zur Zeit Bergmanns (um 1780) umfasste der Reagentiensatz“ des Analytikers schon Lackmus, Veilchen- und Galläpfelsaft, Schwefelsäure, Oxalsäure, Pottasche, Kalkwasser, Höllenstein, Bleizucker und Spiritus.
Im 19. Jahrhundert entwickelte sich schließlich nach Entdeckung immer weiterer Elemente ein für Laien bald kaum noch überschaubares Repertoire an Nachweismethoden und Nachweisreaktionen. Und um zu verhindern, dass bestimmte Stoffe gezielte Nachweisreaktionen (durch Färbungen, Trübungen usw.) störten, entwickelten Chemiker schließlich ein Trennsystem: Sie trennten mit Hilfe bestimmter Fällungsmittel (Gruppenreagentien) die nachzuweisenden Metallsalze (Kationen) in Gruppen von Niederschlägen und Lösungen auf - der klassisch-nasschemische Kationentrenngang entstand. Dieser basierte auf der Basis von Ausfällungen und Säure-Base-Reaktionen und dem methodisch immer gezielteren Einsatz immer gleicher, effizienter Fällungs- und Nachweismittel in Laboratorien.
Anorganische Chemie Bearbeiten
Die erste Prüfung einer kleinen Teilmenge des Stoffgemisches erfolgt nach äußeren Eigenschaften wie
- Farbe,
- Beschaffenheit,
- Kristallform,
- eventuell auch Geruch und Geschmack, jedoch ist wegen der möglichen Toxizität der unbekannten Substanzen stark von Geruchs- und Geschmacksproben abzuraten!
und dann werden in Vorproben
- die Löslichkeit,
- das Verhalten beim Erhitzen (Lötrohrprobe),
- die Färbung der Borax- oder Phosphorperle und
- die Flammenfärbung getestet.
Der größere Teil wird dann vollständig in Lösung gebracht, denn fast nur aus Lösungen lassen sich die für einzelne Ionen charakteristischen Färbungen und Niederschläge erzeugen. Schwer lösliche Verbindungen werden mit starken Säuren oder Salzschmelzen aufgeschlossen.
Im Trennungsgang werden die gelösten Ionen mit Hilfe von Reagenzien in Gruppen aufgeteilt, innerhalb derer weitere Trennungen vorgenommen werden, um schließlich die isolierten Ionen mit Spezialreagenzien nachzuweisen (Identifizierungsreaktion, Nachweisreaktion). Diese Gruppen bezeichnet man als Fällungsgruppen. Der Kationentrenngang wird im Chemiestudium in der Reihenfolge von oben nach unten durchführt:
| Gruppenname | Reagenz | abgetrennte Ionen | Vorgehen | Prinzip |
|---|---|---|---|---|
|
N2H4 | Pt2+/4+, Pd2+/4+ | einige Edelmetalle werden durch Hydrazin zu Metall reduziert und können anschließend wieder gelöst und identifiziert werden | |
|
HCl | Ag+, (Pb2+), (Hg1+) | siehe Salzsäuregruppe | Fällung schwerlöslicher Chloride im sauren |
|
H2S in Salz- oder Essigsäurelösung | Cu2+, Sn2+/4+, Cd2+, Hg1+/2+, Pb2+, As3+/5+, Sb3+/5+ | siehe Schwefelwasserstoffgruppe | Fällung schwerlöslicher Sulfide im sauren |
|
C6H12N4 | Fe3+, Al3+, Cr3+ | siehe Ammoniumsulfidgruppe, Urotropin-Gruppe | Fällung schwerlöslicher Hydroxide dreiwertiger Kationen im alkalischen |
|
(NH4)2S | Co2+, Ni2+, Zn2+, Mn2+ | siehe Ammoniumsulfidgruppe | Fällung weiterer schwerlöslicher Sulfide, nun im alkalischen |
|
(NH4)2CO3 | Ba2+, Sr2+, Ca2+ | siehe Ammoniumcarbonatgruppe | Fällung schwerlöslicher Carbonate im alkalischen |
|
direkt, weil viele Ionen durch die bisherigen Fällungen abgetrennt | Na+, NH4+, K+, Mg2+, Li+ | siehe Nachweisreaktionen, Lösliche Gruppe |
Organische Chemie Bearbeiten
In den vergangenen Jahrhunderten haben sich zahlreiche Menschen mit Einzelaspekten der qualitativen organischen Analyse beschäftigt. Auf diese Weise wurden Spezialreagenzien zum Nachweis verschiedener bedeutender funktioneller Gruppen entwickelt, z. B. Fehlingsche Lösung, Lucas-Probe u. v. a.
Die ersten umfassenden Trennungs- und Analysengänge für die qualitative organische Analyse wurden im 20.Jahrhundert verfasst. Das Schema eines solchen Analysenganges sieht wie folgt aus:
- Trennung des unbekannten Analysengemisches durch Säulenchromatographie, Destillation oder Fällung bzw. Extraktion (Ether-Trennungsgang)
- Bestimmung der physikalischen Eigenschaften der einzelnen Substanzen, also Aggregatzustand, Farbe, Geruch, Siede-/Schmelzpunkt, Brechzahl, pH-Wert der Reinsubstanz und in wässriger Lösung. Unter Zuhilfenahme bestimmter Literatur oder Software, z. B. Beilstein, kann dann die Auswahl der möglichen Stoffe hier schon stark eingegrenzt werden.
- Elementaranalyse durch Natrium-Aufschluss ermöglicht den Nachweis von Stickstoff (Lassaigne-Probe), Schwefel (Bleisulfid-Fällung) und Halogenen (Fällung mit Silbernitrat) in der ursprünglichen Substanz.
- Nachweis funktioneller Gruppen, z. B. Alkohol oder Amin, durch charakteristische Reaktionen
- Eindeutige Identifizierung durch Herstellung eines charakteristischen Derivates
Unterdrückung von Nebenreaktionen Bearbeiten
Oft wird bei Nachweisreaktion neben dem Nachweismittel ein zusätzliches Reagenz zugegeben, welches in der Probe Stoffe beseitigt, die die Nachweisreaktion stören. Die Unterdrückung von Nebenreaktionen kann in vielfältiger Weise geschehen. Die wichtigsten Methoden sind:
Oxidation (Redoxreaktion) Bearbeiten
Wenn z. B. eine Vorprobe auf Acetat durchgeführt wird, um die Anwesenheit von Acetationen in einer unbekannten Probe zu beweisen, dann verreibt man diese Substanz in einem Mörser mit Kaliumhydrogensulfat. Dieser Stoff reagiert mit Acetationen zu Essigsäure, die sich durch ihren Geruch verrät:
- Acetat wird durch Hydrogensulfat protoniert. Es entsteht Essigsäure und Sulfat.

Diese Reaktion wird gestört, wenn eine Probe zusätzlich Sulfidionen aufweist: In diesem Fall entsteht aus Sulfid und Hydrogensulfat das nach faulen Eiern riechende Giftgas Schwefelwasserstoff: Eine Geruchsprobe sollte in diesem Fall natürlich unterbleiben. Zur Beseitigung gibt man zusätzlich etwas Wasserstoffperoxid hinzu: Es oxidiert das störende Sulfid zum geruchlosen Sulfat.
Fällungsmittel (Fällungsreaktion) Bearbeiten
_reactions.JPG)
Viele Schwermetallionen stören Nachweise für Anionen, indem sie mit dem Nachweismittel Farbreaktionen eingehen. Zur Entstörung wird eine Salzprobe daher mit Soda (Natriumcarbonat) und Wasser aufgekocht und filtriert. Sodalösung ist basisch, da Wasser mit Carbonat reagiert:
- Dissoziationsgleichgewicht des Carbonations in Wasser (Säure-Base-Reaktion)

Das Filtrat (Sodaauszug) enthält daher Carbonat- und Hydroxidionen. Störende Schwermetallionen bilden in dieser Lösung Niederschläge (Carbonate und Hydroxide). Das Filtrat wird als Sodaauszug bezeichnet und enthält die Anionen als Natriumsalze. Diese können nun störungsfrei nachgewiesen werden.
Auch im Kationentrenngang wird mit Fällungsmitteln gearbeitet, um störende Kationen voneinander zu trennen - in erster Linie mit Schwefelwasserstoff, welcher Sulfide ausfällt (vgl. Abbildung und siehe unter Salzsäuregruppe, Schwefelwasserstoffgruppe, Ammoniumsulfidgruppe, Ammoniumcarbonatgruppe).
Verdrängung (Säure-Base-Reaktion) Bearbeiten
_reactions.JPG)
Starke Säuren verdrängen schwache Säuren aus ihren Salzen (s. o.). Wenn man Nachweise für Anionen wie z. B. Sulfat durchführt, setzt man hierzu einen Sodaauszug von der Probe ein. Dieser enthält jedoch vom Soda her Carbonat-Ionen (s.o.). Carbonat reagiert wie Sulfat mit dem Nachweismitel Bariumchloridlösung zu einem weißen Niederschlag (es entsteht Bariumcarbonat, weiß, welches einen positiven Sulfatnachweis vortäuscht, denn bei diesem entsteht Bariumsulfat, Malerweiß):

Zum Sulfat-Nachweis wird Salzsäure zugegeben: Sie verdrängt das Carbonat-Ion durch Bildung von Kohlensäure (Kohlendioxid und Wasser; Säure-Base-Reaktion):

Wenn im salzsauren Sodaauszug der Probelösung noch immer ein weißer Niederschlag bei Bariumchloridzugabe auftritt, so muss dieser vom (Barium-)Sulfat her kommen (Fällungsreaktion).
Maskierung (Komplexbildungsreaktion) Bearbeiten
-Ionen_und_Thiocyanat.JPG)
Eine andere Methode ist es, das störende Ion zu „maskieren“. Wenn eine Probe z. B. Kobalt-Ionen enthält, dann lassen sich diese mit dem Nachweismittel Ammoniumthiocyanat und Pentanol (Amylalkohol) nachweisen: Beim Schütteln der Reaktionsmischung entsteht ein in Pentanol blau löslicher Kobalt-Thiocyanat-Komplex (vgl. unter Nachweise für Kationen):
- Cobalt-Kationen reagieren im wässrigen Milieu bei Zugabe von Thiocyanat-Ionen zum pinken Pentaaquathiocyanatocobalt(II)-Komplex, der sich mit blauer Farbe in Pentanol löst.
![{\displaystyle \mathrm {Co^{2+}+SCN^{-}+5\ H_{2}O\longrightarrow [Co(H_{2}O)_{5}(SCN)]^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/96edafdb26bd782483e7a66e128e25544361bb58)
Enthält die Probe jedoch zusätzlich Eisensalze, so überdeckt der bei dieser Nachweisreaktion entstehende, tiefrote Eisen-Thiocyanat-Komplex die blaue Farbe – Eisen stört den Nachweis (Komplexbildungsreaktion):
- Eisen(III)-Ionen und Thiocyanat-Ionen reagieren in einem wässrigen Milieu zum blutroten Pentaaquathiocyanatoferrat(III)-Komplex.
![{\displaystyle \mathrm {Fe^{3+}+\ SCN^{-}+5\ H_{2}O\longrightarrow [Fe(SCN)(H_{2}O)_{5}]^{2+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/b06dd1440c76583922289bb01afc8baeac9e56ed)
Zur Maskierung gibt man beim Nachweis festes Natriumfluorid hinzu: Es reagiert zu einem farblosen, reaktionsunfähigen Hexafluoroferrat-Komplex – das Eisen wurde „maskiert“.
Moderne Bearbeiten
Heutzutage sind qualitative Schnellanalysen mit spezifisch empfindlichen Reagenzien üblich. Auch hat die Instrumentelle Analytik wesentlich mehr Gewicht, selbst für sehr einfache qualitative Fragestellungen. Schwierigere Fragestellungen etwa aus der organischen oder der Biochemie werden meist durch Chromatografie und spektroskopische Methoden gelöst.
Dennoch widmen sich die ersten Semester des Chemiestudiums intensiv dem Kationentrenngang und seinen Nachweisreaktionen, weil er wichtige Stoffkenntnisse vermittelt.
Siehe auch Bearbeiten
Weblinks Bearbeiten
(Grundlagen der Nachweisreaktionen in der Anorganik sowie der Chemie allgemein)
- Anleitung zur qualitativen chemischen Analyse in der Google-Buchsuche Von C. Remigius Fresenius, 9. Auflage, Veröffentlicht von F. Vieweg und sohn, 1856
Literatur Bearbeiten
- Küster Thiel 105. Auflage, "Rechentafeln für die Chemische Analytik", Berlin / New York 2003
- Gerhart Jander: Einführung in das anorganisch-chemische Praktikum. S. Hirzel Verlag, Stuttgart 1990 (in 13. Aufl.), ISBN 3-7776-0477-1
- Udo R. Kunze, Georg Schwedt: Grundlagen der qualitativen und quantitativen Analyse, 5. überarbeitete Auflage, Wiley-VCH, Weinheim, 2002 ISBN 3-527-30858-X
- Michael Wächter: Stoffe, Teilchen, Reaktionen. Verlag Handwerk und Technik, Hamburg 2000, S.154-169 ISBN 3-582-01235-2
- Bertram Schmidkonz: Praktikum Anorganische Analyse. Verlag Harri Deutsch, Frankfurt 2002, ISBN 3-8171-1671-3
Quantitative Analyse Bearbeiten
Eine quantitative Analyse ist ein chemisches und/oder physikalisches Verfahren, bei der es um die Beantwortung der Frage geht, wie viel von einem Stoff in einer gegebenen Probe vorhanden ist.
Alle Methoden lassen sich einteilen in die "klassischen" und die physikalischen Analysemethoden.
Klassische Methoden Bearbeiten
Bei den klassischen Methoden geht es um chemische Umsetzungen, nach denen eine Massen-, Stoffmengen- und Volumenbestimmung vorgenommen wird. Über stöchiometrische Umrechnungsfaktoren lassen sich dann die Massen der einzelnen Elemente bestimmen. Aufgrund der Messungstypen hat man deshalb die folgenden Unterkategorien:
- Gravimetrie oder Gewichtsanalyse
- Volumetrie (Titrimetrie) oder Maßanalyse
Physikalische Methoden Bearbeiten
Bei den physikalischen Methoden geht es um die Messung von konzentrationsabhängigen physikalischen Eigenschaften aus denen eine Konzentration oder Masse berechnet werden kann.
- Elektroanalytische Methoden:
- Konduktometrie (Messung der Leitfähigkeit einer Elektrolytlösung)
- Spektroskopische Methoden nutzen die Wechselwirkung elektromagnetischer Strahlung mit den Molekülen der Probe.
- kernphysikalische Methoden
Physikalisch-chemische Methoden Bearbeiten
Zu den Methoden, bei denen sowohl chemische Reaktionen als auch physikalische Vorgänge eine Rolle spielen gehören:
- Chromatographie mit den Untergruppen:
- Massenspektrometrie mit den Einsatzgebieten
- Elektrochemische Analysemethoden:
- Elektrogravimetrie (Messung des Gewichtes, der elektrolytischen Abscheidung)
- Potentiometrie (Messung der Potenzialänderung)
- Amperometrie (Messung des Elektrolysestroms bei konstantem Potenzial)
- Coulometrie (Messung der benötigten elektrischen Ladung)
- Voltammetrie (Messung von Strom bei bekannten Potenzial)
- Polarographie (Voltammetrie an einer tropfenden Quecksilberelektrode)
- 2D-PAGE (zweidimensionale Gelelektrophorese)
Weblinks Bearbeiten
- Anleitung zur quantitativen chemischen Analyse in der Google-Buchsuche Von C. Remigius Fresenius, 5. Auflage, Veröffentlicht von F. Vieweg und sohn, 1866
Strukturaufklärung Bearbeiten
Strukturaufklärung, auch Strukturanalyse, bezeichnet die Aufklärung der Zusammensetzung chemischer Verbindungen. Chemische Stoffe können sich hinsichtlich der räumlichen Anordnung der einzelnen Atome in ihren Molekülen unterscheiden, auch wenn ihre atomare Zusammensetzung gleich ist (Isomerie). Die Strukturanalytik umfasst als Teilgebiet der Analytischen Chemie chemische und physikalische Methoden zur Aufklärung der chemischen Struktur von Substanzen.
Auch die Strukturchemie, Festkörperchemie, Festkörperphysik und Kristallographie, sowie Werkstoffprüfung und Metallografie beschäftigen sich mit der Aufklärung und Beschreibung von Strukturen, der räumlichen Anordnung von Atomen, Molekülen und Ionen in Feststoffen. In der Oberflächenchemie und -physik ist die Struktur einer Oberfläche von besonderer Bedeutung, da an einem Phasenübergang oft besondere Effekte entstehen.
Chemische Methoden Bearbeiten
Einige funktionelle Gruppen in organischen Molekülen lassen sich mit einfachen chemischen Nachweisreaktionen finden:
- Alkohole: Iodoformprobe
- Aldehyde: Fehlingsche Probe, Schiff's Reagenz, Tollensprobe, Nylanders Reagenz
- Carbonsäuren: Esterbildung mit Geruchsprobe
- C–C-Mehrfachbindungen: Elektrophile Addition von Brom oder Iod (Entfärbung)
- Organische Halogenide: Beilsteinprobe
Die einzelnen Ergebnisse dieser Reaktionen sind häufig nicht als endgültige Nachweis zu verstehen, da manche der Proben nicht vollkommen spezifisch sind oder in Gegenwart bestimmter anderer funktioneller Gruppen versagen. Meist bringt erst die Kombination mehrerer Prüfmethoden Gewissheit über die Struktur der untersuchten Verbindung.
Instrumentelle Methoden Bearbeiten
- Magnetische Kernspinresonanzspektroskopie NMR
- Massenspektrometrie
- Infrarotspektroskopie
- Ramanspektroskopie
- Röntgenstrukturanalyse
- Elementaranalyse
"Kleine Moleküle" Bearbeiten
Durch die Elementaranalyse lässt sich die Zusammensetzung, das heißt der Anteil von Atomen der einzelnen Elemente an einem Molekül, an einer chemischen Verbindung feststellen. Das reicht bei organischen Molekülen jedoch meist noch nicht aus, um eine Strukturformel des Moleküls zeichnen zu können. Um zusätzliche Informationen über die Topologie des Moleküls zu erhalten, stehen eine Reihe spektroskopischer Methoden zur Verfügung.
Dazu gehören:
- NMR: liefert Information über benachbarte Wasserstoffatome, im Molekül relative Abstände der Wasserstoffatome, sowie begrenzte Information über die Verknüpfung der Atome durch deren chemische Verschiebung
- Massenspektrometrie: Zeigt die Gesamtmasse des Moleküls und abhängig von der eingesetzten Technik die Masse von Fragmenten, in die ein Molekül während der Massenspektrometrie zerfällt.
- Infrarot-Spektroskopie: lässt Rückschlüsse über die Existenz bestimmter funktioneller Gruppen im Molekül zu
- Einkristall-Röntgenstruktur-Analyse: führt zu einem dreidimensionalem Modell aller schwereren Atome (Wasserstoffatome werden nur sehr schlecht abgebildet)
Insbesondere ist es oft nötig die Stereochemie einer neu synthetisierten chiralen Substanz zu bestimmen. Für diese Aufgabe kommen von den oben genannten Spektroskopiemethoden nur die Röntgenstruktur-Analyse und in einigen Fällen die NMR-Spektroskopie in Frage.
Bevor diese Techniken bekannt waren, konnte nur eine relative Stereochemie durch Zurückführen der noch nicht charakterisierten Substanz mittels chemischer Reaktionen auf bereits charakterisierte Substanzen bestimmt werden, dies hatte vor allem für die Konfiguration der Zucker eine große Bedeutung.
Biologische Makromoleküle Bearbeiten
Die Strukturaufklärung von Proteinen und DNA heutzutage unterscheidet sich von der kleiner Moleküle, da die Primärstruktur, d.h. die Verknüpfung der einzelnen Atome bereits bekannt ist. Das Interesse gilt hier im allgemeinen der Faltung (auch Proteinstruktur), d.h. der genauen räumlichen Anordnung der Atome im Molekül.
Von den oben genannten Techniken werden für die Aufklärung der räumlichen Struktur von Biomacromlekülen nur die Röntgen-Strukturanalyse und NMR eingesetzt.
Für die Röntgenstrukturanalyse ist es nötig, Einkristalle der Biomacromoleküle in ausreichender Größe zu erhalten. Dies ist oft nur mittels vieler unterschiedlicher Kristallationsversuche möglich, oft werden auch gar keine Kristalle erhalten (zum Beispiel weil das Protein flexible Bereiche aufweist). Der Erhalt von Kristallen kann so Monate bis Jahre in Anspruch nehmen. Sind die Kristalle vorhanden, lassen sich jedoch die entsprechenden Strukturen anhand der aufgenommenen Beugungsmuster normalerweiser innerhalb von Tagen oder Wochen erhalten.
Die Strukturaufklärung mittels NMR analysiert die Biomacromoleküle direkt in Lösung. Es muss daher nicht befürchtet werden, dass die erhaltenen Strukturen durch die Einbettung in ein Kristallgitter und die dadurch zusätzlich auf das Molekül einwirkenden Kräfte verfälscht sind. Durch NMR zugänglich sind jedoch nur Atome mit einem magnetischen Moment (einer ungeraden Spinquantenzahl) des Atomkerns. Insbesondere ist dies Wasserstoff und das natürlich in Kohlenstoff zu 1% neben C12 vorkommende C13 und Phosphor P31 (in DNA und RNA). Um mehr Informationen, auch über andere Atomarten erhalten zu können müssen Moleküle verwendet werden, in denen NMR-taugliche Isotope wie C13 oder N15 angereichert wurden.
Die Analyse von zwei oder dreidimensionalen NMR-Spektren kann die folgenden Informationen über die Substanz liefern:
- Abstände zwischen zwei Wasserstoff-Atomkernen (Protonen) durch den NOE (Kern-Overhauser-Effekt, NOESY-Spektren)
- Orientierung von N15-H Bindungen durch dipolare Restkopplungen
- Diederwinkel durch Bestimmung der skalaren Kopplung in eindimensionalen oder COSY Spektren
Proteine Bearbeiten
Die Strukturaufklärung ist bei Proteinen von Interesse, da nur bei einer von mehreren möglichen Faltungen das Protein in der Lage ist als ein Enzym zu wirken.
DNA und RNA Bearbeiten
Die erste Strukturaufklärung von DNA geht auf Röntgenstrukturaufklärung durch Rosalind Franklin zurück. Ihre Röntgenbeugungsdiagramme lieferten die wesentlichen Hinweise auf die Struktur der DNA, welche im Jahre 1953 von James Watson und Francis Crick veröffentlicht wurde.
Die erste hochaufgelöste Struktur eines DNA-Duplex in B-Konformation, das sogenannte Dickerson-Dodecamer wurde im Jahre 1981 von Drew, Dickerson et al. veröffentlicht. Die Koordinaten dieses Dodecamers sind in der Brookhaven Protein Data Bank unter dem Kürzel 1BNA zugänglich. Es gilt als ein Prototyp für die Struktur von "normaler" DNA in B Konformation und wurde inzwischen in zahlreichen weiteren Studien verfeinert oder als Referenz verwendet.
Bei der Strukturaufklärung von DNA heute, ist oft die Art der Anlagerung von DNA an ein Protein oder eines organischen Moleküls (zum Beispiel eines Arzneimittels) an die DNA von Interesse. Dies gilt insbesondere für chemisch modifizierte DNA, die in Forschung und Analytik eingesetzt wird. Zudem kann DNA Triplexe, Quatruplexe und Haarnadelstrukturen ausbilden.
Die strukturelle Vielfalt von RNA ist generell größer, als die von DNA. Das bedeutet, dass RNA in größerem Umfang als DNA komplexe Strukturen ausbildet, wie zum Beispiel in t-RNA oder snRNA.
Chemische Verbindung Bearbeiten
Als chemische Verbindung bezeichnet man einen Reinstoff, der aus zwei oder mehr verschiedenen chemischen Elementen besteht, die – im Gegensatz zu Gemischen – in einem festen Atomanzahl- und daher auch Massenverhältnis zueinander stehen. Charakteristisch für jede chemische Verbindung ist ihre eindeutige Chemische Struktur. Oft nicht eindeutig ist die Summenformel, mit der man unter Verwendung der molaren Masse beispielsweise die Menge an Produkten einer chemischen Reaktion errechnen kann (in der Stöchiometrie, mit Hilfe eines Reaktionsschemas). Isomere chemische Verbindungen besitzen dieselbe Summenformel, aber eine unterschiedliche Molekülstruktur.
Arten von chemischen Verbindungen Bearbeiten
Grob unterscheidet man bei den über 17 Millionen bekannten chemischen Verbindungen ionische, d. h. salzartige Verbindungen sowie Komplexe, metallische sowie molekulare Verbindungen. Auch die Unterteilung anorganisch/organisch ist grundlegend, wobei als „organisch“ die Kohlenstoff-Verbindungen bezeichnet werden (mit wenigen Ausnahmen). Anorganische Verbindungen gibt es etwa eine halbe Million, organische Verbindungen über 16 Millionen.
Grundsätzlich gibt es also im Hinblick auf die Art der Bindung zwischen den beteiligten Elementen vier Arten von chemischen Verbindungen:
- Molekulare Verbindungen (in der Regel aus einem nichtmetallischen Element und einem oder mehreren weiteren Nichtmetallen)
- Ionische Verbindungen (in der Regel aus einem Metall und einem oder mehreren Nichtmetallen)
- Metallische Verbindungen (aus Metallen)
- Komplexe Verbindungen (aus Metall-Kationen und Ionen oder Molekülen)
Eine genauere Unterscheidung von Verbindungen und deren Zuordnung zu einem dieser vier Typen kann mit Hilfe der Elektronegativitäts-Differenz der an der Bindung beteiligten Elemente vorgenommen werden. Zudem gibt es Übergangsformen zwischen den vier o. g. Idealtypen.
Molekulare Verbindungen Bearbeiten

Molekulare Verbindungen entstehen aus Nichtmetall und Nichtmetall – sie sind Nichtleiter (Isolatoren) mit zumeist relativ niedrigem Siedepunkt (diamantartige oder kunststoffartige Verbindungen mit Riesenmolekülen ausgenommen).
Die kleinsten Teilchen molekularer Verbindungen sind neutrale Atomverbände (Moleküle). Diese bestehen aus zwei Atomen (zum Beispiel im Kohlenmonoxid, CO), aus drei Atomen (zum Beispiel im Kohlendioxid) oder auch aus einigen Tausend oder Zehntausend Atomen (Riesenmoleküle, Polymere, zum Beispiel im Kunststoff Polyethylen oder im Erbgut-Molekül Desoxyribonukleinsäure DNS). Die Atome in den Molekülen sind über Atombindungen verknüpft, das heißt, dass die miteinander verbundenen Atome Paare von Außenelektronen gemeinsam nutzen (Elektronenpaarbindung).
Beispiele für molekulare Verbindungen sind neben Wasser auch Methangas, Zucker, Kohlensäure, Polyethylen usw.
Ionische Verbindungen Bearbeiten

USGOV.jpg)
Ionische Verbindungen (Salze) bestehen aus Kationen und Anionen. Sie sind oft salzartig:
- spröde,
- kristallförmig
- von hohem Schmelz- und Siedepunkt und
- elektrisch leitfähig nur in Schmelze oder Lösung.
Die Ionen entstehen bei der Reaktion von Metall- und Nichtmetallatomen dadurch, dass die Metallatome Elektronen abgeben (Oxidation), die dann von den Nichtmetallatomen aufgenommen werden (Reduktion). Die so gebildeten Metall-Kationen und Nichtmetall-Anionen vereinigen sich auf Grund der elektrischen Anziehungskräfte zu Ionenkristallen. Nach den Nichtmetallen unterscheidet man bei ionischen Verbindungen zum Beispiel Oxide (Sauerstoff als Anion), Sulfide (mit Schwefel), Fluoride, Chloride, Bromide, Iodide, Nitride (mit Stickstoff), Carbide (mit Kohlenstoff), Hydride (mit Wasserstoff) usw. Oft kommt Sauerstoff als drittes Element hinzu; man spricht dann von Sulfaten, Chloraten, Nitraten, Carbonaten usw.
Beispiele für Ionenverbindungen sind Eisen(III)-oxid (dem Rost ähnlich), Pyrit (Eisensulfid), Natriumchlorid (Kochsalz) und Kalziumsulfat (Gips).
Intermetallische Verbindung Bearbeiten
Intermetallische Verbindung (umgangssprachlich oft auch als Legierungen bezeichnet) entstehen aus Metall und Metall – sie sind:
- elektrisch leitfähig
- gut verformbar
- glänzend und
- gute Wärmeleiter
- bei Zimmertemperatur meistens fest.

Die Vereinigung von unterschiedlichen Metallen zu Legierungen kann – ins Besondere beim Zusammengießen von Metallschmelzen – in beliebigen Mengenverhältnissen erfolgen.
Wenn sich jedoch „intermetallische Verbindungen“ bilden, so sind in ihnen die beteiligten Elemente nur in ganz bestimmten Mengenverhältnissen enthalten („intermetallische Phase“, „stöchiometrische Zusammensetzung“, s. unter Stöchiometrie).
Beispiele für Legierungen sind Bronze (aus Kupfer und Zinn), Messing (Kupfer mit Zink) und Kupfernickel (als Münzmetall). Beispiele für intermetallische Verbindungen sind die zwischen Magnesium und Germanium (Formel: Mg2Ge), das Al2Cu, das Magnesiumsilicid Mg2Si, die Bronze Cu4Sn sowie das Zementit Fe3C (aus Eisen und Kohlenstoff, wobei dieser sich wie ein Metall verhält) und WC (Wolframcarbid).
Komplexe Bearbeiten

Verbindungen höherer Ordnung (Komplexe) entstehen bei einer Komplexbildungsreaktion zumeist aus Buntmetallkation und Molekülen mit freien Elektronenpaaren (Liganden). Sie sind oft auffallend farbig.
Beispiele: Der rote Blutfarbstoff Hämoglobin aus Eisen-II-Ionen und Eiweißmolekülen und der tiefblaue Kupfertetrammin-Komplex aus Kupfer-II-Ionen und Ammoniak.
Binäre, ternäre und quaternäre Verbindungen Bearbeiten
Chemische Verbindungen lassen sich auch über die Anzahl der beteiligten chemischen Elemente einteilen.
Binäre Verbindungen setzen sich aus zwei verschiedenen Elementen zusammen und haben die allgemeine Formel AxBy. Chlorwasserstoff (HCl), Natriumfluorid (NaF) und Wasser (H2O) setzen sich jeweils aus zwei Elementen zusammen und sind damit alle binäre Verbindungen. Die Verbindungen können molekulare, ionische (Salze) oder intermetallische Verbindungen sein.
Analog dazu setzen sich ternäre Verbindungen aus drei verschiedenen Elementen zusammen, wie zum Beispiel Natriumcarbonat (Na2CO3), das sich aus Natrium, Kohlenstoff und Sauerstoff zusammensetzt.
Eine quaternäre Verbindung ist beispielsweise Kaliumhydrogencarbonat (KHCO3).
Der Begriff primäre Verbindung macht hingegen keinen Sinn, da definitionsgemäß chemische Verbindungen aus mindestens zwei Elementen zusammengesetzt sein müssen. Unterschiedliche Stoffe aus nur einem Element werden Modifikationen genannt. Gelegentlich wird auch der Begriff Elementverbindung verwendet.
Organische Verbindungen Bearbeiten
Molekulare Verbindungen, in denen Kohlenstoff in Verbindung mit Wasserstoff enthalten ist, werden als „organisch“ bezeichnet. Sie bilden den weitaus größten Teil aller chemischen Verbindungen und leiten sich vom Methangas, der Gruppe der Alkane bzw. den Kohlenwasserstoffen ab. Im Kohlenwasserstoffgerüst organischer Verbindungen befinden sich oft weitere Atomgruppen, die die Eigenschaften der organischen Verbindung beeinflussen.
Dem Kohlenstoffgerüst entsprechend werden organische Verbindungen unterteilt in:
- aliphatische Kohlenwasserstoffe (Aliphaten, hierunter acyclische Kohlenwasserstoffe, gesättigte (Alkane), ungesättigte (Alkene und Alkine) und cyclische Kohlenwasserstoffe),
- aromatische Kohlenwasserstoffe (Aromaten, unterteilt in einfache Aromaten und kondensierte Aromaten),
- Heterocyclen sowie
- biochemische Verbindungen (Alkaloide, Aminosäuren, Kohlenhydrate, Proteine, Steroide, Terpene, Vitamine usw.)
Den funktionellen Gruppe entsprechend teilt man organische Verbindungen ein in
- Sauerstoff- und Hydroxyverbindungen (Alkohole, Aldehyde, Ester, Ether, Ketone, Carbonsäuren usw.),
- Stickstoffverbindungen (Amine, Amide, Nitroverbindungen, Nitrile),
- Schwefelverbindungen (Alkanthiole, Sulfide und Disulfide, Ester der Schwefelsäure, Sulfone, Sulfoxide, Thionamide, Thiolester, Thiosäure),
- Phosphorverbindungen (Phosphate, Phosphine),
- Metallorganische Verbindungen.
Siehe auch Bearbeiten
- Organische Verbindung
- GHS = Global harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien
Weblinks Bearbeiten
Gemisch Bearbeiten

Unter einem Gemisch (Stoffgemisch) versteht man einen Stoff, der mindestens aus zwei Reinstoffen besteht.
Von einem Gemenge spricht man meist bei granulen (Haufwerk, Schüttgut) oder lebenden Komponenten (Samen), die sich nur miteinander vermengen, aber nicht homogen mischen können, ohne abzusterben oder funktionsunfähig zu werden.
Physikalische Chemie Bearbeiten
In der Mischung sind die Ausgangsstoffe unverändert enthalten. Die Ausgangsstoffe werden dabei oft unkenntlich, weil die Mischung andere physikalische Eigenschaften aufweist als jeder isolierte Ausgangsstoff. Beim Mischen entsteht jedoch kein neuer Stoff.
Die spezifischen Eigenschaften wie zum Beispiel Dichte, Siedepunkt oder Farbe sind vom Mischungsverhältnis (Massenverhältnis) der Komponenten abhängig.
- In der Metallurgie wird ein geschmolzenes Gemenge unterschiedlicher Reinsubstanzen letztendlich auch als Legierung bezeichnet.
- In anderem Zusammenhang spricht man von Vermischung oder auch Konglomerat.
- Kolloide sind eine Zwischenform homogener und heterogener Gemische. In diesen Flüssigkeiten sind Feststoffe vermischt, die allerdings in sehr kleinen Phasen von wenigen Molekülen vorkommen und sich deshalb ähnlich wie Lösungen (homogen) verhalten.
Will man Gemische in ihre Reinstoffe auftrennen, so nutzt man die unterschiedlichen physikalischen Eigenschaften aus. Daraus ergibt sich die Auswahl der jeweiligen Trennmethode.
Homogene und heterogene Gemische nach Aggregatzustand Bearbeiten
Die verschiedenen Arten der Gemische, welche nach den Aggregatzuständen der vermischten Stoffe unterschieden werden, lassen sich in die zwei Gruppen unterordnen:
- Heterogene Gemische (Dispersionen) sind nicht vollends vermischt, da die Reinstoffe in klar abgegrenzten Phasen vorliegen, also mehrphasig sind.
- Homogene Gemische sind auf molekularer Ebene vermischte Reinstoffe, also einphasig
Folgende Arten von Gemischen gibt es (rot=homogen, gelb=heterogen):
| Gemisch | fest | flüssig | gasförmig |
| in fest | Legierung, wie Bronze | Schwamm, z. B. vollgesogener Badeschwamm | leerer Schwamm, Hartschaum |
| Gemenge i.e.S., wie Granit | |||
| Haufwerk, wie Kies | |||
| in flüssig | Lösung, wie Wein | ||
| Suspension, wie Schlamm | Emulsion, wie Milch | Schaum | |
| in gasförmig | Aerosol (Oberbegriff) | Gasgemisch | |
| Rauch, Staub, wie Zigarettenrauch | Dampf, Nebel | ||
Arten von Gemischen Bearbeiten
Gemische und Gemenge lassen sich nach dem Phasenzustand, in dem sie vorliegen, unterscheiden. Wenn dabei fest, flüssig und gasförmig zu Grunde gelegt wird, zeigt sich, dass in allen für das menschliche Dasein relevanten Bereichen Gemische und Gemenge weit häufiger von Bedeutung sind als Reinstoffe.
Feststoffgemische Bearbeiten
Feststoffgemische können natürlichen Ursprungs sein oder artifizieller Art (von Menschen geschaffen). Leicht zu definieren scheinen die natürlich vorkommenden Gemische, denen man als örtliche, regionale oder globale Bestandteile der Erdoberfläche begegnet. Beispiele für die Vielfältigkeit solcher Gemische sind Lava, Dolomit oder Granit. Wichtig ist das zur Entstehung Gesagte, denn es zeigt sich, dass alle uns bekannten Feststoffe erst zu solchen geworden sind und sich aus einer im Urzustand heißen und gasförmigen Phase im Zuge derer Abkühlung über flüssig zu zähflüssig und breiig erst in sehr langen erdgeschichtlichen Zeiträumen zu Feststoffen gewandelt haben. Beispiel: Granit ist ein Gemisch oder Gemenge aus Quarz, Feldspat, Glimmer und Hornblende.
Flüssige Gemische Bearbeiten
Natürliche flüssige Gemische sind Laven oder kohlensäurehaltigen Mineralquellen. Bei flüssigen Gemischen ist menschlicher Einfluss bereits seit historischer Zeit nachweisbar. Beispiel: „Griechisches Feuer“, vermutlich ein Gemisch auf Erdölbasis mit Erdpech, Schwefel und Salpeter[1], das bereits von Kaiser Konstantin I. im vierten Jahrhundert als Seekampfmittel genutzt wurde. In modernerer Form begegnet man einer vergleichbaren Mischung in Form des im Zweiten Weltkriegs erfundenen oder wiederentdeckten Molotow-Cocktails, einer Brandflasche, gefüllt mit einem Gemisch aus Benzin und weißem Phosphor.
Gasförmige Gemische Bearbeiten
Das bekannteste gasförmige Gemisch ist unsere Atemluft mit Anteilen an Sauerstoff, Stickstoff, Kohlenstoffdioxid und Helium.
Verfahrenstechnik Bearbeiten
Makrovermischung und Mikrovermischung Bearbeiten
In der chemischen Reaktionstechnik unterscheidet man den Zustand der Vermischung der Reaktionsmasse in einem Reaktor:
- Makrovermischung: das ist alles, was man 'mit dem bloßen Auge erkennen könnte', die Makrovermischung im Reaktor wird charakterisiert durch das Verweilzeitspektrum, das z. B. experimentell durch 'Tracermessungen' erhalten werden kann.
- Mikrovermischung: diese ist ein Charakteristikum des Fluids:
- vollständige Mikrovermischung = molekulardisperses Fluid (Beispiel: Kochsalzlösung)
- vollständige 'Nichtmikrovermischung' = vollständige Segregation = Fluid mit einer Dispersion mikroskopisch kleiner abgeschlossener Fluidelemente (Beispiel: Milch/Emulsion).
Im Englischen und auch bei einigen deutschen Schulen ist die Begriffsdefinition etwas verschieden: engl. macromixing und macrofluid = segregiertes Fluid ; micromixing und microfluid = molekulardisperses Fluid. Für den Begriff 'Makrovermischung', wie er im vorliegenden definiert wurde, wird dann 'contacting pattern' verwendet.
Maßbegriffe von Gemischen Bearbeiten
Anteil, Konzentration oder Gehalt ist der materieller Anteil eines Stoffes an einem Gemisch in Bezug auf Masse, Volumen oder Stoffmenge:
- Massenanteil, Volumenanteil, Stoffmengenanteil
- Volumenkonzentration, Massenkonzentration, Stoffmengenkonzentration
- Molalität
Konkrete Beispiele:
- Salzgehalt, Salinität
Literatur Bearbeiten
- „Herder-Lexikon Geologie und Mineralogie“, Verlag Herder KG, Freiburg im Breisgau 1972, ISBN 3-451-16452-3.
Siehe auch Bearbeiten
- Bestandteil, Vermischung – zu den rechtlichen Belangen des Mischens
- Mischphase (Thermodynamik) – eine homogene Phase, welche aus mehreren Stoffen besteht
- Mischungsaufgaben – eine Untergruppe mathematischer Problemstellungen
Weblinks Bearbeiten
Analysenprobe Bearbeiten
Als Analysenprobe (meist nur Probe oder Stoffprobe genannt) wird in der analytischen Chemie die Gesamtheit des zu untersuchenden Materials bezeichnet. Dieses Material kann als chemischer Stoff oder Stoffgemisch gasförmig (wie eine Autoabgasprobe, eine Geruchsprobe), flüssig (wie Trinkwasserproben, Urinproben), fest (wie eine Gesteinsprobe) oder ein Gemisch unterschiedlicher Aggregatzustände (beispielsweise eine feuchte Bodenprobe) sein.
Analyt, Matrix und Analyselösung Bearbeiten
Der Analyt oder die Analyten sind diejenigen in einer Probe enthaltenen Stoffe, über die bei einer chemischen Analyse eine Aussage getroffen werden soll, d. h. welche analysiert werden sollen. Als Matrix werden diejenigen Bestandteile einer Probe bezeichnet, die nicht analysiert werden. Soll zum Beispiel bestimmt werden, wie viel Chrom ein Nagel enthält, dann ist der Nagel die Probe, das Chrom der Analyt. Eisen, Kohlenstoff und sämtliche weitere eventuell vorhandenen Stoffe bilden zusammen die Matrix. Die Matrix kann unter Umständen die Analyse deutlich erschweren, etwa dann, wenn der Analyt nur einen sehr kleinen Anteil der Probe ausmacht oder die Matrix das Analyseverfahren stört.
Analyselösungen werden in der Chemie verwendet, um Stoffgemische (Proben) zu untersuchen, die von Natur aus nicht flüssig sind, aber mit einem Gerät untersucht werden müssen, das nur flüssige Medien verarbeiten kann. Dazu wird eine definierte Menge der Probe in einem Lösungsmittel, meist Wasser oder Ethanol, gelöst. Die so entstandene Analyselösung wird in das Messgerät gefüllt. Viele Messgeräte sind nur für einen bestimmten Messbereich ausgelegt, so dass es mitunter nötig ist, die Analyselösung weiter zu verdünnen, um sie untersuchen zu können. In diesem Fall muss dann das Messergebnis mit dem Verdünnungsfaktor multipliziert werden, um die reale Konzentration zu ermitteln. Diese Analyselösungen werden etwa für Analysen mittels Atomabsorptionsspektrometer (AAS) oder bei der Radiocarbon-Messung benötigt sowie für alle Arten der nasschemischen Analyse.
Siehe auch Bearbeiten
Nachweisreaktion Bearbeiten
Ein Nachweis ist eine Methode der Analytischen Chemie, die dazu dient, eine Stoffprobe (den Analyten) qualitativ zu untersuchen. Davon zu unterscheiden sind quantitative Bestimmungsmethoden und Methoden der Strukturanalytik.

-Ionen1.jpg)
Die Nachweisreaktion ist eine chemische Reaktion, die das Vorhandensein eines Analyten anzeigt. Die Bilder rechts zeigen beispielsweise eine Niederschlagsbildung im Reagenzglas als Nachweisreaktion für Chlorid-Anionen mit Hilfe von Silbersalzlösung und Ammoniakwasser, darunter zwei Nachweise von Kupfersalzen als Kupfertetramminkomplex (tiefblaue Lösung, Komplexbildungsreaktion) und als Kupferhexacyanoferrat-II (brauner Niederschlag, Fällungsreaktion). Weitere als typische Nachweisreaktionen nutzbare Arten von Stoffumwandlungen sind Redoxreaktionen und Säure-Base-Reaktionen.
Mit dem Nachweis (der Nachweisreaktion) kann eine Probe ohne oder mit relativ einfachen apparativen Mitteln untersucht werden auf:
- in ihr enthaltene Einzelkomponenten (qualitativ),
- deren ungefähre Menge oder Konzentration (halbquantitativ) sowie
- auf strukturelle Besonderheiten, z. B. funktionelle Gruppen
- Spezies, in denen ein Element vorliegt (z. B. Chlor als Chlorid oder Hypochlorit oder elementar)
So werden chemische Elemente, eventuell vorhandener Ionen und funktionelle Gruppen mit Hilfe vieler „Schnelltests“ (Teststreifen oder nasschemische Nachweisreaktionen) in der Probe identifiziert. Von zentraler Bedeutung ist dabei neben diversen Messmethoden vor allem die Sinneswahrnehmung, während zur Konzentrationsanalytik und Strukturanalytik (in Forschung, Produktion (Analytik) und Chemieunterricht) Methoden der instrumentellen Analytik eingesetzt werden. Hierzu zählen z. B. instrumentelle Bestimmungsmethoden der Chromatographie, Spektrometrie, Photometrie, Osmometrie, Refraktometrie, Volumetrie, Gravimetrie und elektroanalytische Methoden.
Methodologie Bearbeiten
Die Methoden umfassen Fällungsreaktionen, Redoxreaktionen, Verdrängungsreaktionen, Komplexbildungsreaktionen und Flammproben. Gegebenenfalls ist die Probe vor Durchführung der Nachweisreaktion aufzubereiten oder von störenden Begleitstoffen zu reinigen.
In der anorganisch-analytischen Chemie geschieht das qualitative Nachweisen von Stoffen in Stoffproben z. B. in Form der Durchführung des Kationentrennganges (vgl. unter Qualitative Analyse und im folgenden Artikelabschnitt).
Anwendungsgebiete und Geschichte Bearbeiten
Quantitative Bestimmungen von Stoffen werden oft mit ähnlichen Nachweisreaktionen durchgeführt, zielen aber darauf ab, Gehaltsangaben für die zuvor qualitativ nachgewiesenen Stoffe zu ermitteln (vgl. unter: Quantitative Nachweise). Diese kommen oft nur in Spuren vor (<1 %), knapp oberhalb der Grenzkonzentration (GK) oder der Erfassungsgrenze (EG) der Nachweisreaktion, so dass physikalische Analysemethoden eingesetzt werden müssen (Gaschromatografie, Atomabsorptionsspektroskopie usw.). So lassen sich heutzutage auch Spurenstoffe im ppb-Bereich erfassen (1 ppb = 1:109; siehe unter: Quantitative Analyse, Instrumentelle Analytik, Analytische Chemie).
Qualitative sowie quantitative Nachweise auch von nur in Spuren vorhandenen Stoffen durchführen zu können, war früher von großer Bedeutung in der Chemie. Das Beispiel des Arsens zeigt diese Bedeutung im Hinblick auf die Kriminalistik: Die Marshsche Probe ist eine Nachweisreaktion in der Chemie und Gerichtsmedizin für Arsen, die 1832 von dem englischen Chemiker James Marsh entwickelt wurde. Vor der Entdeckung der Marsh'schen Probe war Arsen ein beliebtes Mordgift, da es sich nur schwer nachweisen ließ. Ein anderer Arsennachweis ist die Bettendorfsche Probe (über Zinn(II)-chlorid).
Durch die Instrumentelle Analytik und ihre Methoden wie z. B. die spektroskopischen Verfahren hat die Bedeutung von Nachweisreaktionen in der Analytik abgenommen. Für die Vermittlung fachspezifischer Inhalten und Methoden sind sie jedoch nach wie vor von didaktischer Bedeutung (vgl. unter: Chemieunterricht).
Nachweisreaktion Bearbeiten
Die Nachweisreaktion ist also vor allem eine Voruntersuchung zur quantitativen Bestimmung oder zur Strukturaufklärung. Sie hat in der Regel die Funktion eines Schnelltests, der gewisse Hinweise zur Proben-Beschaffenheit liefert.
Nachweise von Ionen Bearbeiten
,(III).JPG)
Nachweisreaktionen für Ionen können in Form von Redoxreaktionen, Säure-Base-Reaktionen, Komplexbildungsreaktionen oder Fällungsreaktionen ablaufen.
Einige Salze sind z. B. nur sehr schlecht wasserlöslich. Dies nutzt man zum Nachweis von Ionen durch Ausfällung. Dazu mischt man sowohl eine wässrige Lösung der Untersuchungssubstanz (Probe, Analyt) als auch eine Referenzlösung (Probelösung) mit dem Nachweismittel. Die in der Vergleichslösung enthaltenen Ionen reagieren mit dem Nachweismittel, ebenso die gegebenenfalls in der Probelösung enthaltenen Ionen. Reagiert die Probelösung wie die Vergleichslösung, so ist der Nachweis positiv. So fallen Eisen-II-kationen dann z. B. bei Zugabe von Blutlaugensalzlösungen als schwer wasserlösliches Salz aus.
Man unterscheidet vom Analyten her in der Anorganik:
Nachweise von Gasen Bearbeiten
Nachweisreaktionen existieren nicht nur für Anionen und Kationen, sondern auch für Gase:
Wasserstoff Bearbeiten
Für den Nachweis von Wasserstoff eignet sich die Knallgasprobe. Das unbekannte Gas wird entzündet. Vernimmt man hierbei einen Knall oder ein lautes Pfeifen, handelte es sich um Wasserstoff:

Sauerstoff Bearbeiten
Sauerstoff weist man mit der Glimmspanprobe nach. Ein glimmender Holzspan glüht in einem Gasgemisch mit hohem Sauerstoffanteil deutlich auf.
Kohlenstoffdioxid Bearbeiten

Für den Nachweis von Kohlenstoffdioxid verwendet man eine Calciumhydroxidlösung. Dazu leitet man das Gas in gesättigtes Kalkwasser oder Barytwasser und ein farbloser Feststoff (Calciumcarbonat) fällt aus.


Ammoniak Bearbeiten
Für den Nachweis von Ammoniak verwendet man gasförmigen Chlorwasserstoff bzw. konzentrierte Salzsäure. Dabei entsteht Ammoniumchlorid, der als weißer Nebel ausfällt.

Nachweise für Säuren und Basen Bearbeiten

Ein Säure-Base-Indikator ist ein Stoff, der durch eine Farbveränderung pH-Wert-Änderungen durch Säuren oder Basen anzeigt. Am häufigsten werden Säure-Base-Indikatoren daher bei Titrationen verwendet (siehe unter Säure-Base-Titration).
Säuren und Basen enthalten in wässriger Lösung Oxonium- bzw. Hydroxidionen. Diese lassen sich beispielsweise mit Universalindikator nachweisen. Hierbei ändert der Indikator abhängig vom pH-Wert der Probesubstanz seine Farbe. Andere Säure-Base-Indikatoren sind zum Beispiel Phenolphthalein und Bromthymolblau.
Nachweis von Wasser Bearbeiten
Wasser entsteht zum Beispiel als Kondensat gasförmigen Wasserdampfes oder als Reaktionsprodukt aus der Neutralisation von Säuren und Laugen. Man weist es mit wasserfreiem Kupfer(II)-sulfat nach: wasserfreies, weißes Kupfersulfat färbt sich bei Zugabe von Wasser hellblau. Es entsteht ein Kupfertetraqua-Komplex, bei dem vier Wassermoleküle als Liganden des Zentralions auftreten:
![{\displaystyle \mathrm {CuSO_{4}+4\ H_{2}O\longrightarrow [Cu(H_{2}O)_{4}]^{2+}\ SO_{4}^{2-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/65fc4dd05db7130b18d3a125db4353f6678438d8)
Dabei handelt es sich um eine Komplexbildungsreaktion.
Ein weiterer sehr empfindlicher Wassernachweis auf der Grundlage von Komplexbildung ist die Rosafärbung von blauem (also wasserfreiem) Cobaltchlorid, dies wird zur Herstellung von Wasserteststreifen aus blauem Cobaltchloridpapier genutzt.
Weitere Nachweisverfahren Bearbeiten
Literatur Bearbeiten
- Gerhart Jander, E. Blasius: Einführung in das anorganisch-chemische Praktikum. S. Hirzel Verlag, Stuttgart 2005 (in 15. Aufl.), ISBN 3-7776-1364-9
- Gerhart Jander, E. Blasius: Lehrbuch der analytischen und präparativen anorganischen Chemie. S. Hirzel Verlag, Stuttgart 2002 (in 15. Aufl.), ISBN 3-7776-1146-8
- Michael Wächter: Stoffe, Teilchen, Reaktionen. Verlag Handwerk und Technik,Hamburg 2000, S.154-169 ISBN 3-582-01235-2
- Bertram Schmidkonz: Praktikum Anorganische Analyse. Verlag Harri Deutsch, Frankfurt 2002, ISBN 3-8171-1671-3
- Eberhard Gerdes: Qualitative Anorganische Analyse. Ein Begleiter für Theorie und Praxis. Springer, Berlin 2001 (2. Aufl.), ISBN 3-540-67875-1
- Dr. Thomas Bitter (Redaktion): Elemente Chemie I - Unterrichtswerk für Gymnasium. Ernst Klett Schulbuchverlag GmbH, Stuttgart 1986 (1. Aufl.), ISBN 3-12-759400-3
Weblinks Bearbeiten
Stoffmengenkonzentration Bearbeiten
| Physikalische Größe | |||||||
|---|---|---|---|---|---|---|---|
| Name | Stoffmengenkonzentration(1) | ||||||
| Formelzeichen | c | ||||||
| Abgeleitet von | Stoffmenge je Volumen | ||||||
| |||||||
| Anmerkungen | |||||||
(1) veraltet: Molarität | |||||||
Die Stoffmengenkonzentration (Formelzeichen: c) oder Molarität (veraltete Bezeichnung) ist der Quotient aus der Stoffmenge (n) eines gelösten Stoffes X und dem Volumen (V) der Lösung. Bei Kenntnis der molaren Masse lässt sich aus der Stoffmengenkonzentration die Massenkonzentration oder - mithilfe der Avogadro-Konstante (vgl. auch Loschmidt-Zahl) - die Teilchenzahlkonzentration berechnen.
Geschichtliches Bearbeiten
Früher wurde das Symbol M für die Maßeinheit Mol pro Liter verwendet und auch zusammen mit SI-Vorsätzen benutzt; diese Schreibweise ist jedoch nicht mit dem Internationalen Einheitensystem (SI) verträglich. Nach IUPAC-Empfehlung soll nur die Stoffmengenkonzentration im Englischen mit concentration bezeichnet werden.[2]
Schreibweise Bearbeiten
Die Stoffmengenkonzentration kann in der Einheit Mol pro Liter angegeben werden:
![{\displaystyle c_{X}={\frac {n_{X}}{V}}\qquad [c_{X}]=1{\frac {\rm {mol}}{\rm {l}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ff54a3d42864ac16211da8af0e6ae3ed170eadfa)
Mit einer 2,5-molaren (veraltete Darstellungsweise auch: 2,5 M) wässrigen Schwefelsäure ist eine Lösung gemeint, die 2,5 Mol Schwefelsäure-Moleküle je Liter bei einer bestimmten Temperatur enthält; hierbei ist die übliche Einheit mol/l zugrunde gelegt;
Rechenbeispiele Bearbeiten
In einer 0,8-molaren (0,8 M) Schwefelsäurelösung (molare Masse von Schwefelsäure: 98,08 g/mol) sind pro Liter Lösung 0,8 mol H2SO4 enthalten. Die entsprechende Masse an H2SO4 für ein Volumen V = 3 Liter einer solchen Schwefelsäure beträgt:

Die Teilchenzahl N einer 3-millimolaren (3 mM) Lösung von Rohrzucker in Wasser bei einem Lösungsvolumen V = 2 Kubikzentimeter, wobei NA (Avogadro-Konstante) die Anzahl der Teilchen pro Mol angibt, beträgt:

Siehe auch Bearbeiten
Chemische Struktur Bearbeiten
Die chemische Struktur gibt den Aufbau auf molekularer Ebene eines einheitlichen (homogenen) Stoffes wieder. Zu ihrer Darstellung werden verschiedene chemische Formeln verwendet. Information über die Struktur einer Verbindung ist oft, entsprechend den Regeln der chemischen Nomenklatur, auch in ihrem Namen enthalten. Moderne Datenbanken arbeiten hingegen mit eindeutigen Strukturcodes wie InChI, SMILES oder WLN.
Instrumentelle Verfahren Bearbeiten
Die Strukturaufklärung von Molekülen wird in der Strukturanalytik heutzutage mit verschiedenen instrumentellen Verfahren vorgenommen.
Nachweisgrenze Bearbeiten
Die Nachweisgrenze (englisch limit of detection, LOD, oder lower detection limit, LDL) bezeichnet den extremen Wert eines Messverfahrens, bis zu dem die Messgröße gerade noch zuverlässig nachgewiesen werden kann (Ja/Nein-Entscheidung).
Der Messwert an der Nachweisgrenze hat eine erhöhte Ungenauigkeit, die aber ein vorgegebenes statistisches Konfidenzintervall nicht überschreitet. Messwerte (Sachverhalte), die eine größere Ungenauigkeit aufweisen als das vorgegebene Intervall, liegen außerhalb der Nachweisgrenze und werden im Sinne der Messtechnik als unmessbar bzw. nicht nachweisbar bezeichnet.
Das Kriterium des „zuverlässigen Nachweises“ wird in der Regel bezogen auf die Präzision des Messverfahrens bei einer Nullmessung oder Leermessung. Gemeint ist damit der statistische Fehler oder die Schwankung des Messsignals wenn keine Probe vorhanden ist (z. B. die Standardabweichung von Untergrundsignal oder Blindwert).
Häufig gilt eine Messung als Nachweis, wenn der Messwert mindestens drei Standardabweichungen über der Nullmessung liegt.

- : Mittelwert des Blindwertes
- : Standardabweichung des Blindwertes


Siehe auch Bearbeiten
Gesamter organischer Kohlenstoff Bearbeiten
Der gesamte organische Kohlenstoff oder TOC-Wert (engl.: total organic carbon) ist ein Summenparameter in der Wasser- und Abwasseranalytik und gibt die Summe des gesamten organischen Kohlenstoffs in einer Wasserprobe an. Er ist das Maß für die organische Verunreinigung der Probe.
Zur Ermittlung des TOC-Gehalt wird die Konzentration des gesamten organisch gebundenen Kohlenstoffs im Wasser bestimmt und zumeist mit automatisierten Messverfahren ermittelt.
Saubere Quellwässer weisen einen TOC-Gehalt von 1–2 mg/l auf. Schwach belastete Flüsse und Bäche zeigen Werte um 2–5 mg/l. In mesotrophen Seen werden bereits Werte um 5–10 mg/l erreicht, in produktiven Karpfenteichen typischerweise 15–25 mg/l. In stark verschmutzten Gewässern kann der Wert auf über 100 mg/l steigen. Der TOC dient neben anderen Summenparametern zur Abschätzung der Wassergüte.
Die Methode beruht auf der Oxidation der im Wasser enthaltenen Kohlenstoffverbindungen und der anschließenden Bestimmung des dabei entstandenen CO2 (Kohlenstoffdioxid). Die Oxidation erfolgt üblicherweise durch die thermische Verbrennung oder alternativ über einen UV-Persulfataufschluss der Wasserprobe, die anschließende Detektion des CO2 mittels Infrarotphotometrie.
Die Bestimmung des TOC in der Wasserchemie steht in einer Konkurrenzbeziehung zur Bestimmung des chemischen Sauerstoffbedarfs (CSB). Die Vorteile der TOC-Bestimmung liegen in der erhöhten Genauigkeit, dem geringeren erforderlichen Probevolumen und der besseren Automatisierbarkeit. Ein weiterer Vorteil gegenüber der CSB-Bestimmung ist, dass hier keine schwefelsauren und hoch mit Schwermetallen (Hg, Ag, Cr) belasteten Abwässer anfallen. Nachteil ist der wesentlich größere apparative Aufwand. Der wesentliche Unterschied beider Methoden besteht aber im Gegenstand der Messung: Bei CSB-Bestimmung spielt die mittlere Oxidationsstufe der aktuell vorliegenden Kohlenstoffverbindungen die entscheidende Rolle. Alle weiteren oxidierbaren Wasserinhaltsstoffe werden mit erfasst oder müssen (wie z. B. Chlorid, Cl−) durch besonderen Aufwand maskiert werden. Die TOC-Bestimmung dagegen ist selektiv, es werden ausschließlich Kohlenstoffverbindungen erfasst, unabhängig von der Oxidationsstufe des Kohlenstoffs.
Die Messung des TOC hat sich vor allem bei der Untersuchung von Trinkwasser und Oberflächenwässern durchgesetzt, da in diesen Fällen eine CSB-Bestimmung oftmals zu ungenau wäre. In der Abwasseranalytik ist dagegen, vor allem in Bezug auf die Belastung und/oder Leistung von Kläranlagen, der CSB-Wert in Kombination mit dem BSB-Wert (Biochemischer Sauerstoffbedarf) als Maß für die organische Belastung verbreitet.
Inzwischen ist auch bei der Abwasserabgabe der TOC-Wert als Maß für die organische Belastung zulässig. Die Umrechnung in CSB-/BSB-Angaben ist jedoch problematisch und nur dann einigermaßen zuverlässig, wenn eine konstante Oxydationsstufe in den meisten kommunalen Abwässern angenommen wird. Dagegen gelten für Industrieabwässer unterschiedlicher Branchen auch höchst unterschiedliche Faktoren zur Umrechnung zwischen beiden Größen.
Analysenmethoden Bearbeiten
In der Normung (z. B. DIN EN1484) werden drei verschiedene Verfahren beschrieben, um den TOC-Gehalt zu ermitteln. Damit wird dem Umstand Rechnung getragen, dass einerseits alle relevanten Verbindungen erfasst werden sollen, andererseits eine Störung der Messung durch Matrix-Effekte möglichst vermieden werden soll. Moderne Analysengeräte können nach allen drei Verfahren betrieben werden. Die Auswahl des geeigneten Verfahrens richtet sich dabei nach der Zusammensetzung und Konzentration der Kohlenstoffverbindungen und nach den zu erwartenden Störstoffen.
NDIR-Detektion Bearbeiten
Zur Bestimmung des Kohlenstoffdioxidgehalts im Trägergas (ausgetriebener TIC/TOC) werden meist Nichtdispersive Infrarotsensoren (NDIR) eingesetzt, welche vom Trägergas konstant bzw. reproduzierbar durchströmt werden. Die durch den NDIR-Detektor gemessene Konzentration wird in Abhängigkeit von der Zeit erfasst. Das resultierende Integral aus CO2-Konzentration über die Zeit (oft auch als Peakfläche bezeichnet) ist dabei ein Maß für den aus der Probe freigesetzten Kohlenstoff C.
Verbrennungsmethode (Combustion) Bearbeiten
Bei der Verbrennungsmethode wird die Probe in einem beheizten Reaktor vollständig verbrannt und der in der Probe enthaltene Kohlenstoff zu Kohlenstoffdioxid CO2 oxidiert.
Feste Proben werden bei ca. 900 °C oder höher verbrannt. Für flüssige Proben beträgt die Verbrennungstemperatur üblicherweise ca. 700 bis 1000 °C, wobei höhere Temperaturen den Probenaufschluss fester wie flüssiger Proben prinzipiell begünstigen. Die Verbrennung und vollständige Umsetzung des in der Probe enthaltenen Kohlenstoffs in der Gasphase zu CO2 wird durch Einsatz von Katalysatoren (z. B. Kupferoxid, Cerdioxid oder platinhaltige Katalysatoren) unterstützt.
Die Verbrennungsgase werden mit einem Trägergasstrom (meist synthetische Luft oder reiner Sauerstoff) transportiert, entfeuchtet / getrocknet und zum Detektor geführt. Die Bestimmung des Kohlenstoffdioxids erfolgt in einem nicht dispersiven Infrarotdetektor (NDIR).
Seit Juni 2009 beschreibt der Normenentwurf DIN EN 15936 die Versuchsverfahren zur Bestimmung des TOC-Gehalts in Feststoffen. Der Entwurf soll die Norm DIN EN 13137 ersetzen und beschreibt die Verbrennung in einem separaten Feststoffanalysator sowie eine neue Suspensionsmethode, um Feststoffe in Flüssiganalysatoren zu bestimmen.
Parallel zum TC oder NPOC kann man den TNb (Gesamter gebundener Stickstoff) mit einem separaten, Stickstoffmonoxid-spezifischen Detektor (z. B. Chemosensor bzw. ECD, Chemolumineszenz-Detektor oder IR-Detektor) ermitteln.
Um den TIC zu erhalten, wird bei den Verbrennungsgeräten die Probe (meist in einem separaten, unbeheizten Reaktor) angesäuert und das frei werdende Kohlenstoffdioxid ausgetrieben. Die Bestimmung des Kohlenstoffdioxids erfolgt wiederum im NDIR-Detektor.
Ein häufiges Problem bei der Verbrennungsmethode sind die in vielen Proben enthaltenen Salze sowie Säuren- oder Laugen-Bestandteile, welche zu vorzeitigen Verschleiß hauptsächlich des Verbrennungsrohres (meist Quarzglas) und Inaktivierung des Katalysators führen können. Seitens der Gerätehersteller gibt es verschiedene Konzepte, diese Problematik zum Teil recht wirkungsvoll zu umgehen (Matrixabscheidung).
Nasschemische Methode „UV-Persulfatmethode“ (Wet-Chemical) Bearbeiten
Die Probe wird in ein beheiztes Aufschlussgefäß eingeleitet. Säure wird zugegeben und der anorganische Kohlenstoff (TIC) in Kohlenstoffdioxid umgewandelt. Das Kohlenstoffdioxid wird mit Stickstoff ausgetrieben und im NDIR – Detektor als TIC gemessen. Der Probe wird Persulfat zugegeben, sie wird mit UV-Licht bestrahlt und der organische Kohlenstoff (TOC) im beheizten Reaktor in Kohlenstoffdioxid umgewandelt. Das Kohlenstoffdioxid wird mit Stickstoff ausgetrieben und im NDIR – Detektor als TOC gemessen. Feste Probenbestandteile (auch suspendierte Partikel) können nicht vollständig aufgeschlossen werden. Daher ist diese Methode für partikelhaltige Proben ungeeignet. Auch salzhaltige Proben sind problematisch, da mit zunehmenden Salzgehalt (insbesondere Chlorid) das Persulfat verstärkt Nebenreaktionen eingeht und für die Oxidation des Kohlenstoffs nicht mehr zur Verfügung steht. Dabei besteht die Gefahr von Minderbefunden.
Einsatzbereich der verschiedenen Methoden Bearbeiten
Die nasschemische Methode „UV-Persulfatmethode“ wird wegen der erreichbaren hohen Empfindlichkeit vorzugsweise für den Nachweis kleinster TOC Gehalte beispielsweise in Pharmawässern (Reinstwasser, WFI, …) verwendet, da eine große Probenmenge (bis ca. 20 ml) eingesetzt werden kann. Nachteilig bei dieser Methode ist neben der Nichteignung für partikelhaltige Proben auch, dass einige sehr stabile organische Verbindungen (z. B. Barbitursäure) unter Umständen nicht vollständig aufgeschlossen werden und dabei die Gefahr von Minderbefunden besteht.
Für den Bereich der Umweltanalytik (neben Abwasser auch Trinkwasser und Oberflächenwasser) wird vorzugsweise die Verbrennungsmethode (Injektionsmenge bis ca. 2 ml) eingesetzt. Der Hochtemperaturaufschluss ist eine Methode, die auch schwer oxidierbare Verbindungen und Partikel sicher aufschließt. Moderne Geräte erreichen auch mit der Verbrennungsmethode eine hohe Messempfindlichkeit und können daher als Alternative zur UV-Methode, u. a. für die Analytik von Pharmawässern, verwendet werden.
Rechenverfahren Bearbeiten
Differenzverfahren (TOC = TC − TIC) Bearbeiten
Im Bereich großer TOC-Konzentrationen (z. B. bei kommunalem Abwasser) wird häufig nach dem „Differenzverfahren“ gearbeitet. Dabei wird im ersten Schritt die Gesamtheit aller Kohlenstoffverbindungen („TC“ = Total Carbon) bestimmt und anschließend in einer zweiten Messung der Anteil anorganischer Kohlenstoffverbindungen (z. B. Karbonate) („TIC“ = Total Inorganic Carbon) ermittelt. Durch Subtraktion des TIC vom TC erhält man dann den TOC-Wert.
Direktverfahren (TOC als NPOC) Bearbeiten
Da sowohl der TC als auch der TIC mit einer Messungenauigkeit behaftet sind, führt das Differenzverfahren bei Proben mit einem gegenüber dem TIC kleinen TOC-Anteil oft zu ungenauen Ergebnissen. Im Bereich der Trinkwasseranalytik wird deshalb das so genannte „Direktverfahren“ eingesetzt. Dafür wird die Probe vor der Messung angesäuert, um den anorganischen Kohlenstoffanteil „TIC“ über Kohlensäure in Kohlenstoffdioxid (CO2) umzuwandeln. Mit einem Inertgasstrom wird das entstandene CO2 anschließend aus der Probe ausgeblasen. Dabei gehen allerdings auch flüchtige Säuren (Ameisensäure aus angesäuertem Formiat, Essigsäure aus angesäuertem Acetat etc.) sowie alle leicht flüchtigen organischen Substanzen (z. B. Treibstoffbestandteile) für die Messung verloren. Deshalb wird auch der so erhaltene TOC als „NPOC“ (Non Purgeable Organic Carbon, nicht ausblasbarer organischer Kohlenstoff) bezeichnet. Dennoch ist dieses Verfahren in relativ TIC-reichen (= harten) Wässern das Verfahren der Wahl, das die beste Genauigkeit und Reproduzierbarkeit liefert.
Additionsverfahren Bearbeiten
Leicht flüchtige organische Verbindungen werden beim Direktverfahren nicht mit erfasst. Darf ihr Anteil am TOC nicht vernachlässigt werden, setzt man das „Additionsverfahren“ ein. Dazu wird der ausgeblasene Gasstrom vom CO2 befreit und die enthaltenen flüchtigen Verbindungen anschließend oxidiert und bestimmt. Der so erhaltene „POC“ (Purgeable Organic Carbon) wird dann mit dem NPOC zum TOC addiert.
Literatur Bearbeiten
- U.S. Environmental Protection Agency (EPA). Cincinnati, OH (2009). Method 415.3: Determination of Total Organic Carbon and Specific UV Absorbance at 254 nm in Source Water and Drinking Water. Revision 1.2. Document no. EPA/600/R-09/122.
- Bernie B. Bernard, Heather Bernard, James M. Brooks: Determination of TC, TOC, and TIC in Sediments
Antioxidans Bearbeiten
Ein Antioxidans (Mehrzahl Antioxidantien) ist eine chemische Verbindung, die eine unerwünschte Oxidation anderer Substanzen gezielt verhindert.
Antioxidantien haben große physiologische Bedeutung durch ihre Wirkung als Radikalfänger. Sie inaktivieren im Organismus reaktive Sauerstoffspezies (ROS), deren Vorkommen im Übermaß zu oxidativem Stress führt. Oxidativer Stress gilt als mitverantwortlich für den Alterungsprozess und wird in Zusammenhang mit der Entstehung einer Reihe von Krankheiten gebracht.
Antioxidantien sind ferner von Bedeutung als Zusatzstoffe für verschiedenste Produkte (Lebensmittel, Arzneimittel, Bedarfsgegenstände, Gebrauchsmaterialien) um darin einen - zum Beispiel durch Luftsauerstoff bewirkten - oxidativen Abbau empfindlicher Moleküle zu verhindern. Der oxidative Abbau bestimmter Inhaltsstoffe oder Bestandteile kann sich wertmindernd auswirken dadurch dass Geschmack oder Geruch sich unangenehm verändern (Lebensmittel, Kosmetika), die Wirkung nachlässt (bei Arzneimitteln), schädliche Abbauprodukte entstehen oder physikalische Gebrauchseigenschaften nachlassen (z. B. bei Kunststoffen).
Wirkungsmechanismus Bearbeiten
Nach Art des chemischen Wirkmechanismus werden Antioxidantien in Radikalfänger oder Reduktionsmittel unterschieden. Im weiteren Sinne werden auch Antioxidationssynergisten zu den Antioxidantien gerechnet.
- Radikalfänger
Bei Oxidationsreaktionen zwischen organischen Verbindungen treten vielfach kettenartige Radikalübertragungen auf. Hier werden Stoffe mit sterisch behinderten Phenolgruppen wirksam, die im Ablauf dieser Übertragungen reaktionsträge, stabile Radikale bilden die nicht weiter reagieren, wodurch es zum Abbruch der Reaktionskaskade kommt (Radikalfänger). Zu ihnen zählen natürliche Stoffe wie die Tocopherole und synthetische wie Butylhydroxyanisol (BHA), Butylhydroxytoluol (BHT) und die Gallate. Sie sind wirksam in lipophiler Umgebung.
- Reduktionsmittel
Reduktionsmittel haben ein sehr niedriges Redox-Potential, ihre Schutzwirkung kommt dadurch zustande, dass sie eher oxidiert werden als der zu schützende Stoff. Vertreter sind etwa Ascorbinsäure (-0,04 V bei pH 7 und 25°C), Salze der schwefligen Säure (+0,12 V bei pH 7 und 25 °C) und bestimmte organische schwefelhaltige Verbindungen (z. B. Glutathion, Cystein, Thiomilchsäure), die vorwiegend in hydrophilen Matrices wirksam sind.
- Antioxidationssynergisten
Synergisten unterstützen die Wirkung von Antioxidantien, beispielsweise indem sie verbrauchte Antioxidantien wieder regenerieren. Durch Komplexierung von Metallspuren (Natrium-EDTA[3]) oder Schaffung einen oxidationshemmenden pH-Wertes können Synergisten die antioxidative Wirkung eines Radikalfängers oder Reduktionsmittels verstärken.
Vorkommen Bearbeiten
Natürliche Antioxidantien Bearbeiten
Viele Antioxidantien sind natürlich und endogen vorkommende Stoffe. Im Säugetierorganismus stellt das Glutathion ein sehr wichtiges Antioxidans dar, auch eine antioxidative Aktivität von Harnsäure und Melatonin ist bekannt. Ferner sind Proteine wie Transferrin, Albumin, Coeruloplasmin, Hämopexin und Haptoglobin antioxidativ wirksam. Antioxidative Enzyme, unter denen die wichtigsten die Superoxiddismutase (SOD), die Glutathionperoxidase (GPX) und die Katalase darstellen, sind zur Entgiftung freier Radikale in den Körperzellen ebenfalls von entscheidender Bedeutung. Für ihre enzymatische Aktivität sind Spurenelemente wie Selen, Kupfer, Mangan und Zink wichtig. Als antioxidativ wirksames Coenzym ist Ubichinon-10 zu nennen. Für den menschlichen Organismus essentiell notwendige und antioxidativ wirksame Stoffe wie Ascorbinsäure (Vitamin C), Tocopherol (Vitamin E) und Betacarotin (Provitamin A) können nicht bedarfsdeckend synthetisiert werden und müssen mit der Nahrung zugeführt werden (exogene Antioxidantien). Eine Reihe von Antioxidantien werden als Bestandteil der Muttermilch an den Säugling weiter gegeben um dort ihre Wirkung zu entfalten.
Als sekundäre Pflanzenstoffe kommen Antioxidantien wie Carotinoide und verschiedenste polyphenolische Verbindungen (Flavonoide, Anthocyane, Phytoöstrogene und andere) in zahlreichen Gemüse- und Obstarten, Kräutern, Früchten, Samen etc. vor und auch in daraus hergestellten Lebensmitteln. Ebenfalls pflanzlichen Ursprungs sind Vitamin C und Vitamin E, die aber auch synthetisch oder teilsynthetisch hergestellt werden können.
| Vorkommen natürlicher Antoxidantien | |
|---|---|
| Verbindung(en) | Lebensmittel mit hohem Gehalt[4][5][6] |
| Vitamin C (Ascorbinsäure) | Frisches Obst und Gemüse |
| Vitamin E (Tocopherole, Tocotrienole) | Pflanzenöle |
| Polyphenolische Antioxidantien (Resveratrol, Flavonoide) | Tee, Kaffee, Soja, Obst, Olivenöl, Kakao, Zimt, Oregano, Rotwein |
| Carotinoide (Lycopin, Betacarotin, Lutein) | Obst, Gemüse, Eier.[7] |
Synthetische Antioxidantien Bearbeiten
Zu den künstlichen Antioxidationsmitteln zählen die Gallate, Butylhydroxyanisol (BHA) und Butylhydroxytoluol (BHT). Durch eine synthetische Veresterung der Vitamine Ascorbisäure und Tocopherol wird deren Löslichkeit verändert um das Einsatzgebiet zu erweitern und verarbeitungstechnische Eigenschaften zu verbessern (Ascorbylpalmitat, Ascorbylstearat, Tocopherolacetat).
Antioxidantien in der Ernährung Bearbeiten
Gesundheitlicher Stellenwert Bearbeiten
Freie Radikale sind hochreaktive Sauerstoffverbindungen, die im Körper gebildet werden und in verstärktem Maß durch UV-Strahlung, Schadstoffe in der Luft und Chemikalien entstehen. Ihr Vorkommen im Übermaß (oxidativer Stress) erzeugt Zellschäden und gilt nicht nur als mitverantwortlich für den Alterungsprozess, sondern wird auch in Zusammenhang mit der Entstehung einer Reihe von Krankheiten gebracht. Ein Schutz vor den schädlichen Folgen durch freie Radikale stellt das körpereigene Abwehrsystem dar, in welchem vor allem Radikalfänger antioxidativ wirksam werden. Außer endogen gebildeten Antioxidantien wirken im Abwehrsystem auch solche, die mit der Nahrung zugeführt werden. Eine gesunde Ernährung unter Einbeziehung von mit an antioxidativ wirksamen Stoffen reichen Lebensmitteln gilt als effektive Vorbeugung vor Herz-Kreislauferkrankungen,[8][9] eine Schutzwirkung vor bestimmten Krebsarten wird als möglich erachtet, ist jedoch nicht durch aussagekräftige Studien gesichert.[8][10][11]
Häufigste Lebensmittelquellen Bearbeiten
Nach einer US-amerikanischen Untersuchung aus dem Jahr 2005 stammt der mit Abstand größte der mit der täglichen Nahrung zugeführte Teil physiologischer Antioxidantien aus dem Genussmittel Kaffee. Dies liege weniger daran, dass Kaffee außergewöhnlich große Mengen an Antioxidantien enthält, als vielmehr an der Tatsache, dass die US-Amerikaner zu wenig Obst und Gemüse zu sich nähmen, dafür aber umso mehr Kaffee konsumierten.[12] In Deutschland und einigen anderen europäischen Ländern dürfte die Lage aufgrund der vergleichbaren Ernährungsgewohnheiten ähnlich sein.
| Rang | Quelle | mg/Tag | Rang | Quelle | mg/Tag |
|---|---|---|---|---|---|
| 1 | Kaffee | 1.299 | 6 | Rotwein | 44 |
| 2 | Tee | 294 | 7 | Bier | 42 |
| 3 | Bananen | 76 | 8 | Äpfel | 39 |
| 4 | Trockenbohnen | 72 | 9 | Tomaten | 32 |
| 5 | Mais | 48 | 10 | Kartoffeln | 28 |
Nahrungsergänzung Bearbeiten
Antioxidativ wirksame Substanzen werden in einer Reihe von Nahrungsergänzungsmitteln als „Anti-Aging“-Präparate und zur Krankheitsprävention (z.B. vor Krebs) auf dem Markt angeboten. Die enthaltenen antioxidativen Substanzen kommen auch natürlicherweise in der Nahrung vor, außerdem werden sie vielen Lebensmitteln zugesetzt, sodass in der Regel kein Mangel besteht. Es fehlen belastbare wissenschaftliche Nachweise, dass die Einnahme von Nahrungsergänzungsmitteln – in denen antioxidativ wirksame Substanzen meist isoliert und nicht im Verbund mit natürlichen Begleitstoffe enthalten sind – gesundheitlich vorteilhaft ist.[14][15] Bei bestimmten physiologischen oder pathologischen Zuständen soll sich eine antioxidative Nahrungsergänzung sogar nachteilig auswirken: bei Krebspatienten wurden Wechselwirkungen mit antineoplastischen Behandlungmethoden (Chemotherapie, Strahlentherapie)[16] oder andere schädliche Auswirkungen[17] beschrieben, bei Sportlern wurde in einer 2009 veröffentlichten Studie ein kontraproduktiver Effekt von Vitamin C und E auf den Trainingseffekt gemessen. Diese Antioxidantien unterdrücken den Anstieg von Radikalen im Körper, so dass er sich weniger gut an die Belastung anpasste.[18][19]
Totale antioxidative Kapazität Bearbeiten
Die Bestimmung der totalen antioxidativen Kapazität (total antioxidant capacity, TAC) in Körperflüssigkeiten liefert einen pauschalen Eindruck über die relative antioxidative Aktivität einer biologischen Probe. Es stehen verschiedene Möglichkeiten für die Bestimmung der antioxidativen Kapazität in Körperflüssigkeiten zur Verfügung. Das Grundprinzip all dieser Methoden ist gleich. Die in der biologischen Probe enthaltenen Antioxidantien schützen ein Substrat vor dem durch ein Radikal induzierten oxidativen Angriff. Die Zeitspanne und das Ausmaß, mit der die Probe diese Oxidation verhindert, kann bestimmt werden und wird meist mit Trolox (wasserlösliches Vitamin E-Derivat) oder Vitamin C als Standard verglichen. Je länger es dauert, um ein Substrat zu oxidieren, desto höher ist die antioxidative Kapazität . Durch verschiedene Extraktionen kann man die antioxidative Kapazität von lipidlöslichen und wasserlöslichen Substanzen untersuchen[20] (siehe auch: Trolox Equivalent Antioxidative Capacity, TEAC).
Angesichts der Bedeutung der Antioxidantien bei der Reduzierung der Risiken von chronischen Erkrankungen wurde 2010 in den USA die totale antioxidative Kapazität durch Ernährung und Nahrungsergänzungsmittel bei Erwachsenen untersucht. Dabei wurden Datenbanken des US Department für Landwirtschaft, Daten zu Nahrungsergänzungsmitteln und zum Lebensmittelverzehr von 4391 US-Erwachsenen im Alter von 19+ Jahren ausgewertet. Um die Daten zur Aufnahme von einzelnen antioxidativen Verbindungen zu TAC-Werte zu konvertieren, wurde die Messung des Vitamin-C-Äquivalent (VCE) von 43 antioxidativen Nährstoffen zuvor angewendet. Die tägliche TAC lag durchschnittlich bei 503,3 mg VCE/Tag, davon ca. 75% aufgenommen durch die Nahrung und 25% durch Nahrungsergänzungsmittel.[21]
Technische Verwendung Bearbeiten
In der Industrie werden Antioxidantien als Zusatzstoffe (Additive) benötigt, um die oxidative Degradation von Kunststoffen, Elastomeren und Klebstoffen zu verhindern. Sie dienen außerdem als Stabilisatoren in Treib- und Schmierstoffen. In Kosmetika auf Fettbasis, etwa Lippenstiften und Feuchtigkeitscremes, verhindern sie Ranzigkeit. In Lebensmitteln wirken sie Farb- und Geschmacksverlusten entgegen und verhindern ebenfalls das Ranzigwerden von Fetten.
Obwohl diese Additive nur in sehr geringen Dosen benötigt werden, typischerweise weniger als 0,5 %, beeinflussen ihr Typ, die Menge und Reinheit drastisch die physikalischen Parameter, Verarbeitung, Lebensdauer und oft auch Wirtschaftlichkeit der Endprodukte. Ohne Zugabe von Antioxidantien würden viele Kunststoffe nur kurz überleben. Die meisten würden sogar überhaupt nicht existieren, da viele Plastikartikel nicht ohne irreversible Schäden fabriziert werden könnten. Das Gleiche gilt auch für viele andere organische Materialien.
Der globale Markt für Antioxidantien zur Verwendung in Kunststoffen und Gummi, Treib- und Kraftstoffen, Schmierstoffen, Klebstoffen und Kosmetika hatte im Jahr 2007 ein Volumen von ca. 880.000 Tonnen. Der mengenmäßig größte Anteil entfiel auf Asien, vor Europa und Nordamerika. Mit Antioxidantien wurde 2007 ein Umsatz von etwa 3,7 Mrd. US$ (2,4 Mrd. €) erzielt.[22]
Kunst-, Kraft- und Schmierstoffe Bearbeiten
Es kommen hauptsächlich sterisch gehinderte Amine (hindered amine stabilisers, HAS) aus der Gruppe der Arylamine zum Einsatz und sterisch gehinderte Phenolabkömmlinge, die sich strukturell oft vom Butylhydroxytoluol ableiten (Handelsnamen Irganox, Ethanox, Isonox und andere).
Lebensmittel, Kosmetika, Arzneimittel Bearbeiten
Zulässige Antioxidantien sind in der Zusatzstoff-Zulassungsverordnung und der Kosmetikverordnung geregelt. Es kommen sowohl natürliche als auch synthetische Antioxidantien zum Einsatz.[23][24]
Beispiele für antioxidative Lebensmittelzusatzstoffe sind in der Tabelle angegeben, siehe dazu auch
| E-Nummer | Antioxidans | Zugelassene Verwendung (Beispiele) |
|---|---|---|
| E220–E228 | Schwefeldioxid und Salze der schwefligen Säure | Trockenfrüchte, Wein |
| E300–E302, E304 | Ascorbinsäure, ihre Salze und Fettsäureester | Fruchtsäfte, Konfitüren, Trockenmilchprodukte, Öle und Fette, Obst- und Gemüsekonserven, Backwaren, frische Teigwaren, Fleisch- und Fischerzeugnisse |
| E306–E309 | Tocopherol und seine Ester | pflanzliche Fette und Öle |
| E315, E316 | Isoascorbinsäure und Natriumsalz | Fleisch- und Fischerzeugnisse |
| E310–E312 | Gallate | Bratöl und -fett, Schmalz, Kuchenmischungen, Knabbererzeugnisse, verarbeitete Nüsse, Trockensuppen, Soßen etc. |
| E319 | tert-Butylhydrochinon (TBHQ) | |
| E320 | Butylhydroxyanisol (BHA) | |
| E321 | Butylhydroxytoluol (BHT) | |
| E392 | Rosmarinextrakt (wirksame Inhaltsstoffe insbesondere Carnosol und Carnosolsäure) |
Fette, Öle, Backwaren, Knabbererzeugnisse, Fleisch- und Fischerzeugnisse, Saucen etc. |
| E586 | 4-Hexylresorcin | frische und tiefgefrorene Krebstiere |
Als Lebensmittelzusatz seit 1968 nicht mehr erlaubt ist aufgrund lebertoxischer Wirkungen die Nordihydroguajaretsäure, ein höchst wirksames Antioxidans zur Haltbarmachung von Fetten und Ölen. Sie ist aber weiterhin in kosmetischen Präparaten verwendbar.
Lebensmitteltechnisch und pharmazeutisch gebräuchliche Antioxidationssynergisten sind unter anderem Citronensäure und ihre Salze (E330-E333), Weinsäure und ihre Salze (E334-E337), Phosphorsäure und ihre Salze (E338-E343) und Ethylendiamintetraessigsäure (EDTA) und ihre Salze (Calciumdinatrium-EDTA, E385).
Literatur Bearbeiten
- L. R. Mahoney: Antioxidantien. In: Angewandte Chemie. Band 81, Nr. 15, 1969, S. 555–563, doi:10.1002/ange.19690811503.
- M. H. Carlsen u.a.: The total antioxidant content of more than 3100 foods, beverages, spices, herbs and supplements used worldwide. In: Nutrition Journal 9, 2010, 3. doi:10.1186/1475-2891-9-3 (Open Access unter CC-by-2.0)
Continuous Flow Analysis Bearbeiten
Continuous-Flow Analysis (CFA) bezeichnet eine Analysentechnik, die nasschemische Analyseverfahren automatisch durchführt. Dabei werden die Verfahrenschritte von photometrisch-analytischen Methoden in einem kontinuierliche Fluss von Probe und Reagenzien in der vorgesehenen Reihenfolge abgearbeitet.

Eine solche Analysenmethode und deren zeitlicher Ablauf ist in der Regel auf einem Methoden-Manifold hardwaremäßig aufgebaut, abgestimmt auf den zu messenden Konzentrationsbereich und auf die Art der Probe. Die Zusammensetzung der zu messenden Proben (Probenmatrix) und vorhandene Störkomponenten können für gleiche Bestimmungen einen anwenderspezifischen Aufbau erforderlich machen.
Das CFA Analysensystem saugt flüssige Proben von einem automatischen Probennehmer an, leitet jede Probe zu den vorhandenen Methoden-Manifolds weiter und teilt diese aliquot auf, für die simultane Durchführung aller aufgebauten Bestimmungsmethoden in jeder Probe.
CFA Analysemethoden sind heute in verschiedenen Anwendungsbereichen als ISO Normen Stand der Technik, wie Wasserbeschaffenheit, Bodenbeschaffenheit (Extrakte), Tabak (Extrakte) oder Lebensmittelanalytik. In den DIN EN ISO Normen für die Wasserbeschaffenheit[25] findet sich der Überbegriff Fließanalyse, unter dem die CFA-Technik mit der verwandten FIA-Technik gemeinsam geführt wird.
Anmerkung: Andere Normen verwenden in der deutschen Übersetzung eher unglücklich auch den Begriff „Kontinuierliche Durchflussanalyse“, der als Analyse eines Durchflusses missverstanden werden kann. Die Verwendung unklarer Bezeichnungen erschwert das Auffinden spezifischer Informationen über Suchmaschinen erheblich.
Die CFA-Technik arbeitet mit regelmäßiger Unterteilung (Segmentierung) des kontinuierlichen Reaktionsstromes mittels Gasblasen (Luft, N2). Zur Abgrenzung gegen die FIA-Technik (Fließ-Injections-Analyse, Injections-Fließanalyse) wird CFA verschiedentlich in der Literatur auch als Segmentierte-Fließanalyse[26] SFA bezeichnet. Die Segmentierung in der CFA-Technik ermöglicht die Aneinanderreihung auch aufwändigerer analytischer Verfahrensschritte, ohne dass dadurch die Verschleppung (Dispersion, Carry-Over) zwischen aufeinanderfolgenden Proben unzulässig groß wird. Moderne Microflow[27] CFA Systeme erzielen selbst bei komplizierten Methoden einen Probendurchsatz von 30 bis 40 Proben pro Stunde, auch wenn der Durchlauf vom Probennehmer durch alle Verfahrensschritte bis zum Detektor 10 bis 25 Minuten beträgt. Dies bedeutet, dass der CFA Analysator gleichzeitig 6 bis 12 Proben durch das analytische System leitet, ohne dass eine Probe die nächste Probe unzulässig beeinflusst.
Näheres zur Technik einer einzelnen CFA Methode Bearbeiten

Der Aufbau einer nasschemisch-analytischen Methode am CFA Analysator wird über ein Fließdiagramm dargestellt und für den Anwender dokumentiert. Im gezeigten Bild muss man sich einen konstanten Fluss von links nach rechts vorstellen. Durch den Einsatz einer Vielkanal-Peristaltikpumpe ist das Verhältnis von Probe und kontinuierlich geförderten Reagenzien zueinander immer konstant, unabhängig von der Geschwindigkeit der Pumpe. Die Absolutmenge der einzelnen Lösung ist durch den eingesetzten Pumpenschlauch definiert, die Farbkodierung ist seit über 40 Jahren weltweit etabliert für flowrated Pump Tubes. Im gezeigten Fall einer 12 Position-Peristaltikpumpe für einen CFA Microflow Analysator ergeben die jeweiligen Dosierschläuche die genannte Menge in µl/min (Zahl).
Das Methoden-Manifold (Analytical Manifold) als über die Jahrzehnte etablierter Begriff umfasst dabei alle Bauteile für die Durchführung der analytischen Methodenschritte, welche sich zwischen der Peristaltikpumpe (= Zahlen) und der Messzelle (= Colorimeter) befinden. Dies kann ein Modul mit Injektoren, Misch- und Reaktionsspiralen sein, aber auch ein Inkubator (Heizbad), eine Dialyseeinheit, ein Durchfluss-Mikrodestillations Modul, eine UV-Aufschlusseinheit oder auch eine Hochtemperatur-Hydrolyseeinheit.
Die Abfolge der analytischen Behandlungsstufen des Methoden-Manifolds kann als Reaktionsstrecke bezeichnet werden. Der kontinuierliche Durchfluss wird am Beginn dieser Reaktionsstrecke regelmäßig mittels Gasblasen (Luft, N2) segmentiert, je nach Technik alle 1-2 Sekunden. Die Dosierung von Reagenzien erfolgt in den bereit segmentierten Strom, heißt in die einzelnen vorbeifließenden Segmente an der jeweiligen Dosierstelle nach einer durch den Aufbau festgelegten Zeit. Die regelmäßige Segmentierung einer 60 sec angesaugten Probe alle 1 sec führt nun dazu, dass in diesem Fall (40 Proben/h, 2:1) von jeder Probe prinzipiell eine 60 fache Wiederholungsmessung erfolgt. Man kann sich dabei die CFA Reaktionsstrecke als Förderband vorstellen, mit einem Reaktionsglas jede Sekunde, deren Inhalt in der Folge weitere Lösungen präzise zugesetzt bekommt, die durch eine Matrixabtrennung (Dialyse) laufen, durch eine Aufschlusseinheit, durch eine Reaktionsstufe mit Inkubation, und so weiter. Am Ende der analytischen Durchführung fließen diese Reaktionslösungssegmente kontinuierlich durch eine Messzelle, wobei die Luftsegmentierung zumeist vorher entfernt wird (debubbling).
Das kontinuierliche Signal der photometrischen Detektion zeigt nun als Graphik den Messwert-Verlauf der nacheinander ankommenden Segmente, welche alle unabhängig voneinander und in gleicher Weise die ganze analytische Abfolge durchlaufen haben. Sichtbar wird damit an der Photometergraphik die Reproduzierbarkeit des analytischen Gesamtsystems, im Augenblick und auch für jede Probe einzeln zum Zeitpunkt ihres individuellen Durchlaufs. Diese einzigartige Eigenschaft der CFA Technik, die Stabilität eines analytischen Systems darzustellen und die Plausibilität auch einer einzelnen Messung grundsätzlich nachvollziehbar zu machen, begründet nach Meinung des Autors den jahrzehntelangen ungebrochenen Erfolg der CFA Technik im chemischen Routinelabor. Die zweite Fließanalysetechnik, FIA, bietet als unsegmentierte Technik diese Qualitätsinformationen nicht.
Der Einsatz von CFA Analysatoren im analytischen Labor von heute, erfolgt für Ionenanalytik und Konventionsparameter zumeist als System für die simultane Bestimmung von 2-5 Parametern in jeder Probe (Wahrnehmung des Autors). Bei 50 Proben je Stunde und simultan 4 Parameter, somit 200 Bestimmungen in der Stunde, übertrifft die CFA Technik im Probendurchsatz auch große Discrete Analyser, bei höherer analytischer Sophistication und erheblich geringeren Kosten.
Beispiele Bearbeiten

- Bei gesamt-Cyanid wird die Probe zuerst mit Säure versetzt und mittels UV komplexes Cyanid aufgespalten, darauf wird das Cyanid in Durchfluss destilliert, danach bildet das Destillat mit Reagenzien eine Färbung deren Stärke gemessen wird[28] Der Durchlauf durch das analytische System dauert je nach Bedingungen etwa 15-20 min.
- Bei gesamt-Phosphor wird die Probe zuerst mit Säure versetzt und mittels UV-Licht wird Organophosphor umgesetzt, darauf wird die Probe bei 95°C hydrolysiert zur Umsetzung anorganischer Phosphorverbindungen, das danach vorliegende ortho-Phosphat bildet mit Reagenzien eine Färbung deren Stärke gemessen wird[29] Der Durchlauf durch das analytische System dauert etwa 20-25 min.
- Bei Bodenextrakten werden die Proben für die Bestimmung von Ammonium-[30] oder Nitrat-Stickstoff zuerst über einen Dialysator geleitet, welcher störende Probenmatrix (Färbung, Huminstoffe...) abtrennt, danach reagieren diffundierte Ionen mit Reagenz zu einer Färbung welche gemessen wird. Der Durchlauf bis zum Detektor dauert je nach Methode 6-12 min. Bei einem Probendurchsatz von 60/h befinden sich damit jeweils 6-12 Proben im Durchlauf durch das analytische System
- Bei Düngemittel-Aufschlusslösungen werden die Proben zuerst in einer Verdünnungsschleife am Manifold 1:10 bis 1:20 verdünnt, in der Folge dialysiert und danach mittels Farbreaktion gemessen. Der Durchlauf bis zum Detektor dauert etwa 8-14 min.
Weitere Anwendungen Bearbeiten
Neben der beschriebenen Anwendung der CFA-Technik für die Durchführung photometrischer Analyseverfahren, gibt es auch Anwendungen mit anderen Detektoren wie Fluorimeter, Flammenphotometer oder Sensortechnik. Dabei wird CFA als Transporttechnik verwendet, welche den raschen Transport von Proben nacheinander durch eine Probenvorbereitung ermöglicht, ohne dass unzulässige Konzentrationsverschleppung von einer Probe zur nachfolgenden Probe entsteht.
Geschichte der CFA-Analysentechnik Bearbeiten
Die CFA Technik wurde in den 1950er Jahren von Leonard Skeggs entwickelt und erlebte in der Folge eine jahrzehntelange Blüte als „die“ Standardtechnik in der klinischen Analytik. Unzählige Publikationen über photometrisch, chemische CFA Verfahren existieren von den 1960er Jahren bis in die 1990er. Dabei entwickelte sich die Gerätetechnik vom Autoanalyzer erster Generation weiter zur verbreiteten Makroflow Technik und hin zu kompakten und schnellen Microflow CFA Systemen.
Ende der 1970er Jahre wurde aus dem CFA Prinzip unter Weglassen der Segmentierung die FIA Technik entwickelt, die sich im universitären Bereich für Einzelbestimmungen und einfachere Verfahren gut etabliert hat. Für größere Routinelabors, mit mehreren simultanen Bestimmungen, erheblichem Probendurchsatz und Sicherheit gegen Einflüsse schwankender Probenmatrix bietet auch heute noch die CFA-Technik die passende Lösung. Geringe Kosten für Verbrauchsmaterial, schnelle Abarbeitung von Probenserien und einfaches Qualitätsmanagement bieten Gewähr für weiteren Einsatz dieser Technik in der Zukunft.
Komplexbildungsreaktion Bearbeiten
Eine Komplexbildungsreaktion ist eine chemische Reaktion (Stoffumwandlung und Teil der Komplexreaktionen) aus dem Bereich der Komplexchemie, bei der ein Metall-Kation mit Molekülen oder Ionen reagiert, die als Lewis-Basen ihre freien Elektronenpaare zur Bildung einer koordinativen Bindung mit dem Kation zur Verfügung stellen. Das Molekül oder Ion wird dabei als Ligand bezeichnet. Komplexbildungsreaktionen sind oft durch Farbumschläge gekennzeichnet. In der Summenformel sowie im Reaktionsschema wird der Komplex durch eckige Klammern angedeutet, in denen vorne das Zentralatom wie z. B. ein Kupferkation und in runden Innenklammen die Liganden stehen (Beispiel: [Cu(NH3)4]2+).
Beispiele für Komplexbildungsreaktionen Bearbeiten
Bildung des Hexaaquakupfer-Komplexes Bearbeiten
Wasserfreies, weißes Kupfersulfat färbt sich bei Zugabe von Wasser hellblau – eine Nachweisreaktion für Wasser. Es entsteht ein Aqua-Komplex des Kupfers, bei dem sechs Wassermoleküle als Liganden des Zentralions auftreten, wobei die beiden axial gelegenen Liganden deutlich weiter vom Zentralion entfernt sind (Jahn-Teller-Effekt):
![{\displaystyle \mathrm {CuSO_{4}+6\ H_{2}O\longrightarrow [Cu(H_{2}O)_{6}]^{2+}+SO_{4}^{2-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fec1a75e061a77a88df2e3e6becd39affc8cc852)
Bildung des Tetraamminkupfer-Komplexes Bearbeiten
Kupfer-II-salze ergeben auch mit Ammoniaklösung bei pH-Werten über 8 tiefblaue Komplexsalz-Lösungen (Kupfertetraamminkomplex [Cu(NH3)4]2+ – auch als Tetraamminkupfer(II) bezeichnet, vgl. Abbildung dieser Nachweisreaktion):
![{\displaystyle \mathrm {Cu^{2+}+4\ NH_{3}\longrightarrow [Cu(NH_{3})_{4}]^{2+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/6d37f99210a906f5833072eb5d2ba02dd91bb36b)
Der Kupfertetraammin-Komplex ist wie der Kupferhexaaquakomplex zweifach positiv geladen, da sich 4 neutrale Moleküle an das Zentralatom binden (streng genommen 4 + 2 Moleküle, nämlich 4 NH3 und 2 H2O; Jahn-Teller-Effekt). Wenn sich jedoch Anionen wie z. B. Chlorid-Anionen an das Zentralatom binden, so kann der Komplex auch negativ geladen sein (z. B. ist der grüne Tetrachlorocuprat-II-Komplex zweifach negativ geladen):
![{\displaystyle \mathrm {Cu^{2+}+4\ Cl^{-}\longrightarrow [Cu(Cl)_{4}]^{2-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/68e32ef65467cf0c29d036fd7e7191c84f6b96f7)
Anionische Komplexe werden andersartig benannt (Metall-Name mit Endung -at am Namensende) und sind von großer Bedeutung auch in der Biochemie.
Bildung des Nickel-Dimethylglyoxim-Komplexes Bearbeiten
Nickelsalzlösung bildet mit einer alkoholischen Dimethylglyoxim-Lösung (DMG) Komplexe. In ammoniakalischer Lösung fällt das himbeerrote Nickel-dimethylglyoxim als Komplex aus:

![{\displaystyle \mathrm {[Ni(C_{4}H_{7}N_{2}O_{2})_{2}]+2\ H^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/830f7b97e6beeeedd65d385f2942bdd372563bae)
Dieser Komplex ist im Unterschied zum Kupfertetrammin- oder Kupfertetraquo-Komplex neutral und in Wasser unlöslich.
Komplexbildung als Gleichgewicht Bearbeiten
Derlei Lewis-Säure-Base-Reaktionen zur Komplexbildung sind Gleichgewichtsreaktionen, für die das Massenwirkungsgesetz aufgestellt werden kann. Die resultierende Gleichgewichts-Konstante nennt man Komplexbildungskonstante. Sie gibt auch an wie stabil der Komplex ist bzw. ob er zur Dissoziation neigt. Ihr reziproker Wert wird als Komplexdissoziationskonstante KD bezeichnet, also KA−1 = KD.
Einordnung und Bedeutung von Komplexbildungsreaktionen Bearbeiten
Neben der Komplexbildungsreaktion gibt es in der Komplexchemie auch Ligandenaustausch- und Dissoziationsreaktionen, bei denen Komplexe erzeugt oder wieder abgebaut werden. Viele Komplexbildungsreaktionen sind in Bereichen wie der Katalyse, der Biochemie, der Farbstoffchemie, der Analytischen Chemie mit ihren Nachweisreaktionen insbes. für Kationen im Kationentrenngang sowie der Chemischen Technologie von großer Bedeutung.
Viele farbige Komplexe dienen ferner der Erforschung von Atom- und Molekülstrukturen (so z. B. der Chrom-Hexamin-Komplex, Abbildung, oder die neuartigen Sandwichkomplexe), von Stoffwechselvorgängen (Hämoglobin bei der Atmung, Chlorophyll bei der Photosynthese,Komplexe mit Eisensulfid-Mineralien bei der chemischen Evolution und der Entstehung des Lebens („Eisen-Schwefel-Welt“, vgl. unter chemische Evolution), viele Enzyme, einige Vitamine), Licht-Materie-Wechselwirkungen (Pigmente und Farbstoffe) und von katalytischen Vorgängen.
Fällungsreaktion Bearbeiten
Fällungsreaktionen nennt man chemische Reaktionen, bei denen die Edukte im Lösungsmittel gelöst vorliegen und mindestens ein Produkt in diesem Lösungsmittel un- oder schwerlöslich ist. Das Produkt mit schlechter Löslichkeit fällt aus, die Ausfällung wird allgemein Niederschlag genannt. In Reaktionsgleichungen wird das Ausfallen eines Stoffes mit einem ↓ oder einem (s) für solid hinter der Summenformel des Stoffs gekennzeichnet.
Fällungsmittel Bearbeiten
Fällungsmittel sind Stoffe oder Stoffgemische, welche die Ausfällung gelöster Stoffe zu unlöslichen Feststoffen bewirken (dem Niederschlag).[31]
Als Fällungsmittel im Kationentrenngang der Analytischen Chemie dienen z. B. Salzsäure, Schwefelwasserstoff, Ammoniumsulfid und Ammoniumcarbonat, da sie unlösliche Schwermetallchloride, -sulfide und -carbonate bilden.
Fällungsmittel haben einen besonderen Stellenwert bei der Abwasserreinigung (siehe auch Fällung, Flockung, Flockungsmittel) zur Entfernung von Sulfiden oder Phosphaten.[32]
Beispielreaktionen Bearbeiten
Die Fällung von Bleisulfidniederschlag aus Bleisalzlösung:

(Eine Nachweisreaktion für Sulfidanionen bzw. Blei(II)-kationen)
Die Fällung von Bariumsulfat aus Bariumchloridlösung:
(Eine Synthesereaktion zur Herstellung von „Malerweiß“)
Beispiele von Fällungsreaktionen von Halogenen; X = Cl, Br, I:

Anwendung von Fällungsreaktionen Bearbeiten
- Nachweisreaktion für Ionen
- Durch das Verwenden von spezifischen Fällungsreaktionen ist es möglich, einzelne Bestandteile einer Lösung zu identifizieren. Klassisches Beispiel ist der Kationentrenngang, bei dem Fällungsreaktionen sowohl zur Ionenidentifikation als auch zum Ausfällen von störenden Ionen verwendet werden.
- In der Gravimetrie sind Fällungsreaktionen Grundlage vieler Gehaltsbestimmungen und auch Teile der Volumetrie (speziell die Argentometrie) arbeiten mit Fällungen.
- In der Abwasserreinigung ist die Fällung die übliche Methode, um die Phosphatkonzentration zu senken (Phosphorelimination). Dazu wird Eisen(II)-sulfat, Eisenchlorid, Aluminiumchlorid, Natriumaluminat oder Polyaluminiumchlorid hinzugefügt. Im Prinzip funktionieren alle Metallsalze ähnlich. Jedoch hat jedes einzelne Metallsalz spezielle Vorzüge, die mit örtlichen Gegebenheiten, Anlagenkonstruktion, Abwasserzusammensetzung und dem Reinigungsziel abgestimmt werden müssen. Man unterscheidet bei der Phosphatelimination vier Verfahren, die sich durch die Dosierstellen unterscheiden und unterschiedliche Auswirkungen auf die Reinigungsziele haben.
- Vorfällung: Die Dosierstelle befindet sich im Zulauf der Vorklärung. Wirkung: Die Anlage wird entlastet. Hier ist der Einsatz von 3- und höherwertigen Metallsalzen üblich.
- Simultanfällung: Die Dosierstelle befindet sich im Zulauf der biologischen Stufe oder direkt in der biologische Stufe. Wirkung: größte Effizienz des eingesetzten Fällmittels, gängigste Phosphatfällung, geeignet auch für die kostengünstigeren zweiwertigen Metallsalze, die speziell bei großen Anlagen mit entsprechenden Bedarfsmengen genutzt werden.
- Nachfällung: Die Dosierstelle befindet sich im Zulauf der Nachklärung oder in der gesonderten 3. Reinigungsstufe. Die 3. Reinigungsstufe findet heutzutage kaum noch Verwendung da sie gegenüber der Simultanfällung sehr teuer ist. Überraschenderweise wird im europäischen Ausland diese Phosphat-Reinigungsstufe bei neuen Anlagen wieder gebaut. Wirkung: Gezielte Fällung, die durch eine Phosphatmessung in der biologischen Stufe gesteuert werden kann. Nur 3- und höherwertige Metallsalze möglich.
- Flockungsfiltration: Die Dosierstelle befindet sich im Zulauf der Filtration. Filteranlagen sind nur bei einigen großen Klärwerken vorhanden. Dort wird die Flockenfiltration eingesetzt um beste Ergebnisse zu erreichen und die Ablaufwerte noch einmal zu senken. Üblicherweise wird diese Methode bei Einleitung in Trinkwasserreservoirs verwendet wie z. B. am Bodensee.
- Aufarbeitung von Proben
- Störende Stoffe in einer Analytlösung werden ausgefällt und somit entfernt. Dieses Verfahren wird dann auch Umfällung genannt und stellt im Unterschied zur Umkristallisation eine chemische Reaktion dar. Zum Beispiel entfernt eine Carrez-Klärung Proteine aus einer Lösung.
- Funktionelle Proteomik: alternativmedizinischer Bluttest
Weblinks Bearbeiten
Flammenfärbung Bearbeiten

Die Flammenfärbung, auch Flammprobe genannt, ist eine Methode zur Analyse von chemischen Elementen oder deren Ionen (Nachweisreaktion). Die Methode beruht darauf, dass die Elemente oder Ionen in einer farblosen Flamme Licht spezifischer Wellenlängen abgeben, das für jedes Element charakteristisch ist. Die Flammenfärbung entsteht durch Energieumwandlung von Wärmeenergie zu Strahlungsenergie. Die Umwandlung kommt durch Valenzelektronen zustande, die durch die Wärmeenergie in einen angeregten Zustand gehoben werden und unter der Abgabe von Licht wieder zurückfallen. Stoffe, mit denen Flammenfärbung möglich ist, finden aufgrund dieser Eigenschaft in der Pyrotechnik Anwendung.
Bei der Flammenfärbung wird die Stoffprobe meist einfach auf einem Platindraht oder einem Magnesiastäbchen in die farblose Flamme eines Bunsenbrenners gehalten. Aufgrund der Farbe kann nun auf die Ionen in der Probe rückgeschlossen werden, allerdings überdeckt die sehr intensive gelbe Flammenfärbung des Natriums oft alle anderen Flammenfärbungen. Mit Sicherheit kann nur mit Hilfe eines Spektroskops entschieden werden, welche Elemente in der Probe vorliegen, zumal sich z. B. die Flammenfärbungen von Kalium und Rubidium recht ähnlich sind.
Zu unterscheiden ist die Flammenfärbung von der Lichtabgabe der Edelgase, die auch auf einem angeregten Zustand basiert, welche aber durch Strom, nicht durch eine Flamme herbeigeführt wird.
Erklärung der Flammenfärbung Bearbeiten

Alle Elemente senden bei hohen Temperaturen Licht aus. Doch für Elemente, die eine Flammenfärbung aufweisen, geschieht dies schon bei den Temperaturen, die in einer Flamme herrschen.
Die äußersten Elektronen eines Atoms werden durch Zufuhr von Wärmeenergie (die in diesem Fall durch eine Verbrennung entsteht) auf ein vom Atomkern weiter entferntes, nicht von Elektronen besetztes Energieniveau – in einen angeregten Zustand – gehoben. Diese Elektronen besitzen nun eine höhere potentielle Energie. Die negativ geladenen Elektronen fallen aber meist in Sekundenbruchteilen wieder auf das energieärmere Ausgangs-Energieniveau zurück. Die beim Zurückfallen frei werdende Energie wird als Photon (Lichtteilchen) abgegeben. Man spricht von einem Quant. Es ist durch eine genau definierte Energie und somit auch mit einer einzigen Wellenlänge gekennzeichnet.
Das Zurückfallen der Elektronen auf energieärmere Energieniveaus kann auch stufenweise erfolgen. Bei jedem Zurückfallen dieses Elektrons auf ein energieärmeres Energieniveau gibt es nun Licht einer ganz bestimmten Wellenlänge (Farbe), und damit einer ganz bestimmten Energie, ab.
Farbe der Flammenfärbung Bearbeiten
-
 Antimon, hellblau
Antimon, hellblau -
 Arsen, fahlblau
Arsen, fahlblau -
 Blei, fahlblau
Blei, fahlblau -
 Bor, kräftig grün
Bor, kräftig grün -
 Calcium, ziegelrot
Calcium, ziegelrot -
 Kalium, violett
Kalium, violett -
 Kupfer, grün, auch blau
Kupfer, grün, auch blau -
 Kupfersulfat, stark grün
Kupfersulfat, stark grün -
 Lithium, karminrot
Lithium, karminrot -
 Natrium, natriumgelb
Natrium, natriumgelb -
 Natrium, gelb
Natrium, gelb -
 Strontium, rot
Strontium, rot


Weitere Flammenfärbungen:
- Barium, fahlgrün
- Caesium, hellblauviolett
- Rubidium, rotviolett
- Europium, rot
- Indium, tiefblauviolett
- Selen, blau
- Thallium, grün
- Radium, karminrot
Die freigegebene Lichtenergie hängt von der Differenz der Energieniveaus (ΔE) ab. Diese Differenz ist für jedes Element unterschiedlich. Die Energie der Photonen bestimmt ihre Wellenlänge (λ) und damit die Farbe. So ergibt sich die spezifische Flammenfärbung.

c = Lichtgeschwindigkeit
h = Plancksches Wirkungsquantum
λ = Wellenlänge
ν = Frequenz
Weist ein Element eine spezifische Flammenfärbung auf, dann weisen auch viele Verbindungen seiner Ionen diese Flammenfärbung auf (Beispiel: Bariumsulfat weist eine grünliche Flammenfärbung auf, Bariumphosphat nicht). Sehr viele Elemente senden bei hohen Temperaturen sichtbare Spektrallinien aus. Einige Elemente wurden sogar nach der Farbe ihrer bei der Flammenfärung beobachteten Spektrallinien benannt: Caesium (lateinisch: himmelblau) , Rubidium (lateinisch: dunkelrot) und Indium (indigoblaue Spektrallinie).
Moderne Techniken Bearbeiten
Bessere Möglichkeiten als die klassische Flammenfärbung mit Hilfe des Auges bieten die spektroskopischen Verfahren der Atomspektroskopie, die eine Art Weiterentwicklung dieser mit Hilfe von Messinstrumenten darstellen. Das Auge wird hier durch das Spektrometer ersetzt, welches die Lage der Spektrallinien sehr viel besser auflöst, sowie auch die nicht sichtbaren Bereiche des elektromagnetischen Spektrums je nach Spektroskopieart (z.B. IR- oder UV/VIS-Spektroskopie) zur Analyse nutzt. Außerdem ist es weit besser als das Auge in der Lage, die Stärke der Spektrallinien zu bestimmen, wodurch eine quantitative Analyse möglich wird.
Literatur Bearbeiten
- W. Biltz, W. Fischer, Ausführung qualitativer Analysen anorganischer Stoffe, 16. Auflage, Harri Deutsch, Frankfurt am Main, 1976.
- G. Jander, E. Blasius, Einführung in das anorganisch chemische Grundpraktikum, 14. Auflage, S. Hirzel Verlag, Stuttgart, 1995, ISBN 3-7776-0672-3
Weblinks Bearbeiten
Photometrie Bearbeiten
Mit Photometrie oder Fotometrie (zu altgr. φῶς „Licht“ sowie μετρεῖν „messen“) werden Messverfahren im Wellenlängenbereich des ultravioletten und des sichtbaren Lichtes mit Hilfe eines Photometers bezeichnet.
Teilgebiete Bearbeiten
Die Photometrie ist ursprünglich ein Teilgebiet der Physik beziehungsweise der Chemie, Astronomie und der Photographie, inzwischen aber eine reguläre Ingenieurswissenschaft. Sie wird beispielsweise in der Photovoltaik oder auch bei der Herstellung von Anzeigen für die industrielle Messtechnik zur Qualitätssicherung und Qualitätskontrolle ständig weiterentwickelt. Für die Entwicklung von Optischen Technologien wie der Lasertechnik gehört sie wie die verwandte Kolorimetrie ebenfalls zum Handwerkszeug.
Darüber hinaus findet die Photometrie besonders auch in der (bio-)chemischen und medizinischen Analytik Verwendung. Sie erlaubt den qualitativen und quantitativen Nachweis ebenso wie die Verfolgung der Dynamik chemischer Prozesse von strahlungsabsorbierenden chemischen Verbindungen.
Die Verallgemeinerung der Photometrie auf das gesamte elektromagnetische Spektrum (Radio- bis Gammastrahlung) nennt man Radiometrie.
Transmissionsmessungen Bearbeiten


Absorption und Farbe einer Flüssigkeit oder eines transparenten Festkörpers hängen von der stofflichen Zusammensetzung und der Konzentration ab. Mit der Photometrie werden mithilfe des sichtbaren Lichtes die Konzentrationen von farbigen Lösungen bestimmt.
Bestrahlt man die Lösung eines absorbierenden Stoffes mit Licht, hängt die Absorption von den Transmissionseigenschaften des Stoffes, der Konzentration und der Länge des Lichtweges in der Lösung ab. Diese Gesetzmäßigkeit wird durch das Lambert-Beersche Gesetz beschrieben und ist i.A. wellenlängenabhängig. Das transmittierte Licht wird gemessen. Um den Transmissionsgrad in Abhängigkeit von der Konzentration darzustellen, wird die Transmission in die Extinktion umgerechnet:
Die Extinktion E ist der negative dekadische Logarithmus des Transmissionsgrades T:

Trägt man die Extinktionen bei einer Wellenlänge gegen die Konzentrationen auf, so entsteht eine Gerade. Das Verhältnis zweier Konzentrationen ist dabei gleich dem Verhältnis der entsprechenden Extinktionen:

Man kann so die Extinktion der Lösung mit der unbekannten Konzentrationen messen und daraus die Konzentration der Lösung berechnen:

Um die Konzentration einer Lösung zu messen, wird ein Wellenlängenbereich gewählt, der von den zu bestimmenden Molekülen oder Ionen absorbiert wird, nicht jedoch von anderen Bestandteilen. Manche Substanzen, die keine oder nur geringe Absorption zeigen, können mit chemischen Mitteln in gut absorbierende Substanzen umgewandelt werden. Beispielsweise lässt sich mit Formaldoxim die Konzentration zahlreicher Metallionen photometrisch bestimmen. Licht mit der ausgewählten Wellenlänge wird mit Filtern, Monochromatoren, oder Lasern erzeugt.
Reflexionsmessungen Bearbeiten
Photometrische Untersuchungen betreffen hier vorrangig die Farbbewertung von Oberflächen zur Qualitätssicherung bei der Farbgebung. Es werden kalibrierte, mittels Filtern bei mehreren Wellenlängen messende Photosensoren eingesetzt.
Aus der möglicherweise wellenlängenabhängigen diffusen Reflexion kann auch auf die Oberflächenstruktur geschlossen werden (z. B. DRIFTS).
Bewertung von Lichtquellen Bearbeiten
Die photometrische Bewertung von Lichtquellen bezieht sich auf ihre auf die Hellempfindlichkeitskurve des Auges bezogenen Eigenschaften wie Farbwiedergabeindex, Lichtstrom, Farbtemperatur und Leuchtkraft.
Auch die Abstrahlcharakteristik von Leuchten und Leuchtmitteln (so bei Leuchtdioden) fällt darunter.
Astronomie Bearbeiten
In der Astronomie gibt es weitere photometrische Systeme, die sich nicht an der Empfindlichkeitskurve des Auges anlehnen, sondern an physikalischen Eigenschaften der Sternspektren.
- Die Breitbandphotometrie
- misst die Stärke der Strahlung über einen weiten Wellenlängenbereich. Die gebräuchlichsten Verfahren messen durch drei oder vier Filter (UBV: Ultraviolet, Blue, Visual, oder uvby: ultraviolet, violet, blue, yellow) und bestimmen hieraus die Parameter eines Sterns (Spektraltyp). Die Magnitudendifferenzen der einzelnen Filtermessungen werden als Farben bezeichnet, U-B oder B-V, die oft als Farben-Helligkeits-Diagramm aufgetragen werden (siehe auch Farbindex).
- In der Schmalbandphotometrie
- werden nur Bereiche einzelner Spektrallinien gemessen, um deren Stärken zu bestimmen, ohne ein Spektrum aufzunehmen, was weit aufwändiger wäre. Dies funktioniert jedoch nur bei starken Absorptionslinien und Linienemissionsspektren ohne (starken) kontinuierlichen Anteil wie zum Beispiel die Spektren planetarischer Nebel.
Die Astronomie benutzt aus historischen Gründen als Einheit die Magnitude.
Photometrische Größen Bearbeiten
Die folgenden photometrischen Größen sind aus den zugehörigen radiometrischen Größen abgeleitet. Der Unterschied besteht darin, dass in der Photometrie die Empfindlichkeit des Betrachters mit einbezogen wird, indem die radiometrischen Größen mit der spektralen Hellempfindlichkeitskurve multipliziert werden.
| Größe | SI-Einheit (Zeichen) | Bemerkung | radiometrische Entsprechung |
|---|---|---|---|
| Lichtstrom | Lumen (lm) | Strahlungsleistung einer Lichtquelle, gewichtet mit der Empfindlichkeitskurve | Strahlungsleistung |
| Lichtmenge | Lumensekunde (lms) | Strahlungsenergie einer Lichtquelle, gewichtet mit der Empfindlichkeitskurve | Strahlungsenergie (Strahlungsmenge) |
| Lichtstärke | Candela (cd) | Lichtstrom pro Raumwinkel, gemessen in großer Entfernung von der Lichtquelle; gibt an, wie intensiv eine Lichtquelle in eine bestimmte Richtung leuchtet. Für eine räumlich isotrop strahlende Lichtquelle ist der Lichtstrom gleich der Lichtstärke multipliziert mit , dem vollen Raumwinkel | Strahlstärke |
| Beleuchtungsstärke | Lux (lx) | Lichtstrom pro beleuchteter Fläche; gibt an, wie intensiv die Fläche beleuchtet wird | Bestrahlungsstärke |
| Spezifische Lichtausstrahlung | Lux (lx) | emittierter Lichtstrom, bezogen auf die Größe der Licht abstrahlenden Fläche | Spezifische Ausstrahlung |
| Leuchtdichte | Candela pro Quadratmeter (cd/m²) | Lichtstärke einer Lichtquelle, bezogen auf deren Fläche; diese Größe nimmt der Mensch als Helligkeit einer Licht abstrahlenden Fläche wahr | Strahldichte |

Weitere Größen und Werte:
- Belichtung (Lux mal Sekunde, lx·s), das Produkt aus Beleuchtungsstärke und Zeit
- Mit zunehmender Beleuchtungsstärke sinkt die benötigte Zeit, um eine gleich bleibende Belichtung zu erhalten.
- Lichtausbeute (Lumen pro Watt, lm/W)
- Effizienz der Umsetzung von Leistung in Licht.
- Hellempfindlichkeit (dimensionslos)
- Ähnlichste Farbtemperatur (Kelvin)
Literatur Bearbeiten
- Bergmann, Schaefer: Lehrbuch der Experimentalphysik. Berlin 1990.
- Lange, Zdeněk: Photometrische Analyse. Verlag Chemie, Weinheim 1980, ISBN 3-527-25853-1.
Weblinks Bearbeiten
Titration Bearbeiten

oben: Bürette mit Maßlösung,
unten: Erlenmeyerkolben (besser Titrierkolben) mit Probelösung
Die Titration (Titrimetrie, Volumetrie oder auch Maßanalyse) ist ein Verfahren der quantitativen Analyse in der Chemie. Ein bekannter Stoff, dessen Konzentration unbekannt ist (Probelösung), wird in einer gezielten chemischen Reaktion mit einer Maßlösung, deren Konzentration genau bekannt ist, umgesetzt. Das Volumen der verbrauchten Maßlösung wird dabei gemessen und anhand der Stöchiometrie die unbekannte Konzentration der Probelösung berechnet.[33]
Das Verfahren ist auch mit geringem apparativen Aufwand möglich und wird daher schon früh in der Grundausbildung eingesetzt. Da die Messergebnisse bei optimierten Titrationsverfahren sehr genau sind und sich die Titration gut automatisieren lässt, findet es heute noch breite Anwendung in der chemischen Analytik.
Allgemeines Verfahren Bearbeiten
Mit einer Bürette wird zu einer Probelösung (Titrand) ein Reagenz bekannter Konzentration (die Maßlösung, auch Titrator oder Titrant genannt), hinzugetropft, bis die Äquivalentstoffmenge erreicht ist (auch Äquivalenzpunkt oder Endpunkt genannt). Die Endpunktserkennung kann hierbei vielfältig mit chemischen und physikalischen Methoden erfolgen und unterscheidet auch die verschiedenen Titrationsarten. An der Bürette kann das verbrauchte Volumen abgemessen werden. Vor Beginn der eigentlichen Titration wird der Gehalt der Maßlösung genau bestimmt und ein Korrekturfaktor, der sogenannte Titer ermittelt, um die Genauigkeit der Messung zu erhöhen.
Reaktionstypen Bearbeiten
Titrationen lassen sich nach Typ der chemischen Reaktion unterscheiden.
Säure-Base-Titration Bearbeiten
Bei der Titration kommt es zur Säure-Base-Reaktion. Die Endpunkterkennung kann durch Zusatz von pH-Indikatoren und einen Farbumschlag erfolgen. Es ist auch möglich den pH-Wert mit Elektroden zu messen und den Endpunkt durch Auftragung von pH-Wert und verbrauchter Maßlösung zu ermitteln.
Fällungstitration Bearbeiten
Es werden Fällungsreaktionen zur Bestimmung genutzt. Die Reaktion von Silberionen Ag+ mit Chloridionen Cl− (Argentometrie) zeigen den Endpunkt manchmal durch Zusammenballen des milchigen Niederschlages an (Methoden nach Gay Lussac und Liebig), manchmal unterstützt durch die Zugabe eines Farbstoffes wie Eosin oder Fluorescein (Titration nach Fajans) oder durch Entstehen eines farbigen Produktes wie Eisenrhodanid (nach Titration nach Volhard) oder Silberchromat (Titration nach Mohr).
Ein Sonderfall ist die hydrolytische Fällungstitration, bei der mit einem Alkalisalz einer schwachen Säure titriert wird. En Beispiel hierzu ist die Bestimmung der Gesamthärte mit Kaliumpalmitatlösung, bei der das Palmitation nach Überschreiten des Äquivalenzpunktes mit Wasser hydrolytisch zu Hydroxidionen reagiert.
Komplexometrische Bestimmung Bearbeiten
Die Bestimmung beruht auf Komplexbildungsreaktionen. Dabei können Farbstoffe zugesetzt werden oder die Farbänderung durch Bildung eines Komplexes photometrisch verfolgt werden und somit auch instrumentell bestimmt werden. Weit verbreitet ist die Titration mit EDTA.
Redox-Titration Bearbeiten
In manchen Fällen kann man Redox-Reaktionen zur Bestimmung ausnutzen. Bekannte Verfahren sind die Manganometrie, Iodatometrie, Bromatometrie oder die Cerimetrie, die jeweils nach verwendeten Maßlösung benannt sind.
Phasentransfer Bearbeiten
Die 2-Phasen-Titration nach Epton dient zur Bestimmung von ionischen Tensiden in wässriger Lösung. Der Endpunkt ist der Farbumschlag einer Farbstoffmischung in der organisch-chlorierten Phase.
Polyelektrolyttitration Bearbeiten
Sie dient der Bestimmung des kationischen Bedarfs von Polyelektrolyten. Als Titrant wird bei anionischen Suspensionen PolyDADMAC und bei kationischen KPVS verwendet.
Endpunktserkennung Bearbeiten
- chemische Indikatoren (visuell)
- Farbumschlag von Indikatoren
- Niederschläge (Fällungsreaktionen)
- physikalische Indikatoren (instrumentell)
- potentiometrische Titration beruht auf der sprunghaften Veränderung des elektrochemischen Potenzials
- pH-Wert-Messung mittels Glaselektrode
- Biamperometrie
- Konduktometrische Endpunktbestimmungen beruhen auf Messungen der elektrischen Leitfähigkeit
- Thermometrische bzw. Enthalpische Endpunktsbestimmungen.
Titrationsarten Bearbeiten
Direkte Titration Bearbeiten
Bei der direkten Titration werden Probelösung und Reagenzlösung unmittelbar miteinander umgesetzt. Dabei wird die Probelösung vorgelegt und mit der Reagenzlösung direkt titriert. Bei der inversen Titration wird hingegen eine abgemessene Menge an Reagenzlösung mit der Probelösung titriert.
Indirekte Titration Bearbeiten
Bei der indirekten Titration wird der zu untersuchende Stoff vor der Titration in einer chemischen Reaktion umgesetzt. Der zu bestimmende Stoff wird in einer chemischen Reaktion zu einem genau festgelegten anderen Stoff umgesetzt, der dann titrimetrisch bestimmt wird. Man unterscheidet weiterhin in Rücktitration, bei der die Probelösung mit einem bestimmten Volumen an Reagenzlösung vollständig umgesetzt wird und anschließend der unverbrauchte Teil der Reagenzlösung durch eine Titration bestimmt wird, sowie Substitutionstitration, bei der ein zu bestimmender Stoff zunächst einen anderen Stoff (z.B. Ersatzkation) freisetzt („substituiert“), der dann rücktitriert werden kann.
Spezielle Titrationen Bearbeiten
- Aminzahl
- Bromzahl
- Chemischer Sauerstoffbedarf (CSB)
- Epton Titration
- Esterzahl (EZ; Menge von KOH in mg, erforderlich für die Verseifung von Estern in 1 g Fett)[34]
- Halbtitration zur Bestimmung der Säurekonstante (pKS) einer Säure
- Iodzahl[34]
- Karl-Fischer-Verfahren zur Bestimmung des Wassergehaltes
- Leitfähigkeitstitration
- Neutralisationszahl (NZ; Menge von KOH in mg, erforderlich für die Neutralisation aller freien Säuren in 1 g Fett)
- Ölzahl
- Peroxidzahl (POZ, Menge des als Peroxid gebundenen Sauerstoffs in mval O2 pro kg, der sich leicht als aktiver Sauerstoff abspalten lässt)
- Säurezahl (SZ; Menge von KOH in mg, erforderlich für die Neutralisation der freien organischen Säuren in 1 g Fett)[34]
- Verseifungszahl[34]
- wasserfreie Titration
Automatisierte Titration Bearbeiten

Die verschiedenen Reaktionstypen und Titrationsarten können im automatisierten Laborreaktorsystem realisiert werden. Ein Laborautomatisierungssystem erfasst mit Hilfe einer geeigneten Sonde (pH-Glaselektrode, Leitfähigkeitssonde, Trübungssonde, Farbsonde...) den Zustand und steuert über eine Dosierpumpe die Zugabe der Reagenzlösung. Die zugegebene Menge wird meist durch automatische Messung der Gewichtsabnahme des Reagenz-Vorlagbehälters ermittelt.
Volumetrie ohne Titration Bearbeiten
Eine besonders einfache Art der Volumetrie dient zur Volumenbestimmung eines Gases, indem dieses ein entsprechendes Flüssigkeitsvolumen verdrängt. Zu beachten ist, dass die Flüssigkeit passend zum zu bestimmenden Gas gewählt werden muss. Die Stickstoffbestimmung per Azotometer etwa nutzt dies, indem als Flüssigkeit eine Kaliumhydroxidlösung eingesetzt wird: diese absorbiert bei der Bestimmung ebenfalls entstehendes Kohlenstoffdioxid und Wasser und erlaubt es, das Stickstoffvolumen direkt an einer Maßskala der Azotometerbürette abzulesen.
Siehe auch Bearbeiten
Einzelnachweise Bearbeiten
- ↑ Meyers Konversationslexikon, erschienen in Bibliographisches Institut, Leipzig und Wien, 5. Auflage von 1897, Bd. 7
- ↑ http://www.iupac.org/reports/1993/homann/physical/43.html
- ↑ The European Medicines Agency: Note for Guidance on Inclusion of Antioxidants and Antimicrobial Preservatives in Medicinal Products
- ↑ Beecher G: Overview of dietary flavonoids: nomenclature, occurrence and intake. In: J Nutr. 133. Jahrgang, Nr. 10, 1. Oktober 2003, S. 3248S–3254S, PMID 14519822 (nutrition.org).
- ↑ Antioxidants and Cancer Prevention: Fact Sheet. National Cancer Institute, abgerufen am 27. Februar 2007.
- ↑ Ortega RM: Importance of functional foods in the Mediterranean diet. In: Public Health Nutr. 9. Jahrgang, 8A, 2006, S. 1136–40, doi:10.1017/S1368980007668530, PMID 17378953.
- ↑ Goodrow EF, Wilson TA, Houde SC: Consumption of one egg per day increases serum lutein and zeaxanthin concentrations in older adults without altering serum lipid and lipoprotein cholesterol concentrations. In: J. Nutr. 136. Jahrgang, Nr. 10, Oktober 2006, S. 2519–24, PMID 16988120.
- ↑ a b Deutsche Gesellschaft für Ernährung (DGE): Obst und Gemüse. 29. November 2007. Abgerufen am 4. Mai 2011.
- ↑ Deutsche Gesellschaft für Ernährung (DGE): Gemüse und Obst - Multitalente in Sachen Gesundheitsschutz. 7. Juni 2005. Abgerufen am 4. Mai 2011.
- ↑ Deutsche Krebsgesellschaft: Gesund essen, gesund bleiben. Antioxidantien – wie sie wirken. 2. Oktober 2010. Abgerufen am 4. Mai 2011.
- ↑ Informationsdienst des deutschen Krebsforschungszentrums (DKFZ): Ernährung und Krebsvorbeugung: Kann die Ernährung das Krebsrisiko beeinflussen? 17. April 2007. Abgerufen am 4. Mai 2011.
- ↑ American Chemical Society: Coffee is number one source of antioxidants
- ↑ Coffee is number one source of antioxidants. Abgerufen am 9. Mai 2011
- ↑ Vitamin C- Viel hilft viel?; UGB – Gesundheitsberatung
- ↑ Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ: Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. In: Lancet. 361. Jahrgang, Nr. 9374, Juni 2003, S. 2017–23, doi:10.1016/S0140-6736(03)13637-9, PMID 12814711 (elsevier.com).
- ↑ Antioxidanzien können Krebs-Patienten schaden. Bei: Internisten im Netz vom 8. August 2008
- ↑ Können Vitamine und Mineralstoffe Arteriosklerose vorbeugen?; UGB – Gesundheitsberatung
- ↑ M. Heberer:Vitaminpräparate steigern Diabetes-Risiko. In: Informationsdienst Wissenschaft vom 11. Mai 2009
- ↑ M. Ristow, K. Zarse, A. Oberbach, N. Klöting, M. Birringer, M. Kiehntopf, M. Stumvoll, C. R. Kahn, M. Blüher: Antioxidants prevent health-promoting effects of physical exercise in humans. In: Proceedings of the National Academy of Sciences of the United States of America Band 106, Nummer 21, Mai 2009, S. 8665–8670, ISSN 1091-6490. doi:10.1073/pnas.0903485106. PMID 19433800. PMC 2680430 (freier Volltext).
- ↑ Nicole Unger-Manhart Freie Radikale und Antioxidantien.
- ↑ Yang M et al.: Estimation of total antioxidant capacity from diet and supplements in US adults. The British journal of nutrition 2011 Feb 15:1-11.
- ↑ Marktstudie Antioxidantien von Ceresana Research
- ↑ Antioxidant Ingredients, INCI
- ↑ Zusatzstoff-Zulassungsverordnung - ZZulV
- ↑ Determination of nitrite nitrogen and nitrate nitrogen and the sum of both by flow analysis (CFA) and spectrometric detection, ISO 13395:1996
- ↑ Segmented Flow Analysis, Encyclopedi of Analytical Science, Vol 8, Academic Press
- ↑ The use of Microcontinuous Flow Analysis and FIA in Water Analysis, Straka M., International Laboratory, 09-1990, pg 33
- ↑ Water quality - Determination of total cyanide and free cyanide by continuous flow analysis, ISO 14403:2002(E), First edition 1. März 2002
- ↑ Determination of ortho phosphate and total phosphorus contents by flow analysis, Part 2: Method by continuous flow analysis (CFA), ISO 15681-2
- ↑ Determination of ammonium by flow analysis and spectrometric determination, EN ISO 11732
- ↑ Brockhaus ABC Chemie, VEB F. A. Brockhaus Verlag Leipzig 1965, S. 389−390.
- ↑ Otto-Albrecht Neumüller (Hrsg.): Römpps Chemie Lexikon. Frank’sche Verlagshandlung, Stuttgart 1983, 8. Auflage, ISBN 3-440-04513-7, S. 1232.
- ↑ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8. "titration" doi:10.1351/goldbook.T06387.
- ↑ a b c d Otto-Albrecht Neumüller (Hrsg.): Römpps Chemie Lexikon. Frank’sche Verlagshandlung, Stuttgart 1983, 8. Auflage, ISBN 3-440-04513-7, S. 1265–1270.
Literatur Bearbeiten
- Jander, Jahr: Maßanalyse. 17. Auflage, de Gruyter, Berlin 2009, ISBN 978-3-11-019447-0.
- Leo Gros, Peter A. Bruttel, Marcus von Kloeden: Praktikum der Titration. Metrohm AG.
- Christian Haider: Elektroden in der Potentiometrie. Metrohm AG.
- Peter A. Bruttel: Nicht-wässrige Titrationen von Säuren und Basen mit potentiometrischer Endpunktbestimmung. Metrohm AG.
- Wolfgang Richter, Ursula Tinner: Practical aspects of modern Titration. Metrohm AG.
Weblinks Bearbeiten
Gravimetrie (Chemie) Bearbeiten
Die Gravimetrie ist ein quantitatives Analyseverfahren, bei dem die Messung von Stoffmengen auf der Bestimmung der Masse (Auswaage) beruht. Dabei werden wiederum Fällungsanalyse, Elektrogravimetrie und Thermogravimetrie unterschieden.
Verfahren Bearbeiten
Fällungsanalyse Bearbeiten
Hierbei werden die Ionen bzw. Moleküle in eine Fällungsform gebracht. Die gefällte Verbindung wird abfiltriert. Die Filtration kann entweder in einem Porzellanfiltertiegel bzw. Glassfiltertiegel erfolgen oder in einem Filterpapier (Wobei spezielle aschefreie Filterpapiere zum Einsatz kommen). Anschließend wird der Filterkuchen gewaschen und getrocknet. Falls mit einem Papierfilter filtriert wurde muss dieser noch verascht werden. In manchen Fällen wird für die Fällungsanalyse die Fällungsform durch Glühen in speziell hierfür vorgesehenen Öfen (Muffelöfen) in eine stöchiometrische Wägeform umgewandelt, womit die quantitative Bestimmung der Inhaltsstoffe erfolgen kann.
Mitunter sind Fällungs- und Wägeform identisch. Dies ist insbesondere dann der Fall, wenn der Niederschlag eine eindeutige Stöchiometrie aufweist und beispielsweise keine wechselnden Mengen von Kristallwasser gebunden werden: die Bestimmung von Sulfationen als Bariumsulfat, die Fällung von Nickel mit Diacetyldioxim, die Bestimmung von Kalium mit Natriumtetraphenylborat. Ein Beispiel, bei dem die Fällungsform und die Wägeform nicht identisch sind, ist die unten aufgeführte Bestimmung von Eisen als Eisen(III)oxid.
Elektrogravimetrie Bearbeiten
Die gesuchte Substanz wird bei der Elektrogravimetrie an einer Elektrode abgeschieden und anschließend verwogen.
Thermogravimetrie Bearbeiten
Bei thermogravimetrischen Analyseverfahren wird die Änderung der Stoffmasse in Abhängigkeit von der Temperatur untersucht. Ein einfaches Beispiel dafür sind die gravimetrischen Methoden zur Bestimmung der Feuchte.
Beispiele Bearbeiten
Bestimmung des Eisengehaltes einer Fe(III)-Salzlösung Bearbeiten
Die Eisensalzlösung wird mit Ammoniakwasser versetzt, das ausfallende Hydroxid (Fällungsform) wird abfiltriert und anschließend durch Glühen bis zur Gewichtskonstanz in Eisen(III)-Oxid überführt. Die Masse des Oxids wird durch Auswägen auf der Analysenwaage bestimmt.
(Basische Reaktion von Ammoniak)

(Fällungsform)

(Wägeform)

Die gesuchte Masse a des zu bestimmenden Elementes (in unserem Beispiel Eisen) ist proportional der ausgewogenen Masse A der Wägeform (hier ). Der Proportionalitätsfaktor (Gravimetrischer Faktor) gibt an, zu welchem Anteil die Masse a in der ausgewogenen Masse A enthalten ist.


Aus der ausgewogenen Masse ergibt sich durch die Multiplikation mit einem Faktor die Masse a des zu bestimmenden Elementes.
- bzw. .


enthält zum einen das Verhältnis der Molmassen des zu bestimmenden Elements, , zur Molmasse der Wägeform, . Zusätzlich muss jedoch auch berücksichtigen, „wie oft“ sich eine Formeleinheit der gesuchten Substanz schließlich pro Formeleinheit in der Wägeform wiederfindet. In diesem Beispiel finden sich 2 Eisenatome je Formeleinheit des Wägeproduktes wieder. Dies wird durch den stöchiometrischen Koeffizient k, hier k=2, ausgedrückt:



wobei k der stöchiometrische Koeffizient, die Molekülmasse der gesuchten Substanz und die Molekülmasse der Wägeform ist.
In unserem Beispiel nehmen wir an, wir haben 1,25 g ausgewogen. Wie viel Eisen enthielt ursprünglich unsere Fe(III)-salzlösung?
Einsetzen von
ergibt:
a = m(Fe) = 0,874 g.
Das bedeutet, unsere Fe(III)-Salzlösung enthielt 874 mg Eisen.
Bestimmung von Sulfat Bearbeiten
Die sulfathaltige Probelösung wird mit Salzsäure angesäuert. Unter Rühren wird eine 0.1 M Bariumchloridlösung eingetropft, bis sich am Eintropfpunkt kein Niederschlag mehr bildet. Der Niederschlag wird (zur Ostwaldreifung) auf einem Sandbad über Nacht getempert. Dabei bilden sich auf Kosten der kleinen Kristallite größere Kriställchen, die sich leichter abfiltrieren lassen. Der Niederschlag wird abfiltriert, mit Wasser und Ethanol gewaschen und getrocknet. Anschließend wird der Niederschlag bei 600 °C bis zur Gewichtskonstanz geglüht. Dies dauert im Regelfall nicht länger als 2–3 Stunden.
(Fällungs- und Wägeform)

Bestimmung von Nickel(II) Bearbeiten
Die wässrige Nickel(II)-haltige Probelösung wird tropfenweise mit einer alkoholischen Lösung von Dimethylglyoxim versetzt, bis kein weiterer rötlicher Niederschlag mehr ausfällt. Dann wird das Ethanol verkocht. Anschließend filtriert man über eine Glasfritte ab. Nach waschen des Niederschlages trocknet man im Trockenschrank bis zur Massenkonstanz. wobei Dimethylglyoxim mit H[DMG] abgekürzt wird
![{\displaystyle {\rm {{Ni}^{2+}+2{\rm {{H[DMG]}\rightarrow {\rm {{Ni[DMG]}_{2}\downarrow +2{\rm {{H}^{+}}}}}}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2bf42bedf3d4d0f5b63d2a04853cadfba79cef22)
Gravimetrische Methoden zur Feuchtemessung Bearbeiten
Das am häufigsten verwendete Verfahren zur Messung des Wassergehalts von Materialproben ist die gravimetrische Methode (auch Darr-Wäge-Trocknung). Der Wassergehalt der Materialprobe wird dabei durch den Gewichtsverlust beim Trocknen bestimmt.
Die Materialprobe wird nach der Entnahme luftdicht verpackt und gewogen. Anschließend wird die Probe in einem Trockenofen bei ca. 105 °C getrocknet bis sich Gewichtskonstanz bei aufeinanderfolgenden Wägungen einstellt. Die Trocknungsdauer und -temperatur ist jedoch materialabhängig und in entsprechenden Normen festgelegt. Bei der Trocknung darf kein chemisch gebundenes Wasser freigesetzt werden. Deshalb ist die Trocknungstemperatur von Gips nur 40 °C. Nach dem Trocknen wird die Materialprobe erneut gewogen. Aus der Differenz der Wägungen lässt sich der Wassergehalt der Materialprobe ermitteln.
Der gravimetrische Wassergehalt ergibt sich aus der Masse der feuchten Probe und der Masse der trockenen Probe :




Vorteile Bearbeiten
Die Vorgehensweise bei der Entnahme der Probe und der Trocknungsprozedur ist als Referenzmethode anerkannt. Viele andere Verfahren werden mit dieser Methode verglichen bzw. die indirekten Methoden kalibriert. Vorteile dieser Methode sind die relativ einfache Handhabung und die im Allgemeinen gute Genauigkeit.
Nachteile Bearbeiten
Ein Nachteil des gravimetrischen Verfahrens ist, dass bei der Trocknung organischer Materialien chemische Umwandlungen durch Oxidationsprozesse einsetzen können. Diese können aufgrund der Sauerstoffentnahme die anschließende Wägung zu höheren Werten beeinflussen. Genauso kann es auch zu einer thermischen Zersetzung mit einem daraus resultierenden Gewichtsverlust kommen. So ist in kolloidhaltigen Böden wie Lehm oder Ton die vollständige Entfernung des eingelagerten Wassers nur unter der Zerstörung der Kolloidstruktur möglich.
Aussicht Bearbeiten
Die gravimetrische Methode ist für langfristig angelegte Feldmessungen nicht zu empfehlen. Für Labormessungen ist sie jedoch als Referenzverfahren etabliert. Die langen Trocknungszeiten können durch eine Schnelltrocknung mittels
abgekürzt werden.
Andere direkte Messverfahren zur Bestimmung des Wassergehalts in Böden haben sich nicht durchgesetzt. Dies liegt teilweise an der umständlichen Handhabung, dem erforderlichen Aufwand oder an der geringen Genauigkeit.
Quellen Bearbeiten
- Kupfer, K. 1997 Materialfeuchtemessung: Grundlagen, Messverfahren,Applikationen, Normen, Expert Verlag
- Hübner, C. 1999 „Entwicklung hochfrequenter Meßverfahren zur Boden- und Schneefeuchtebestimmung“, Wissenschaftliche Berichte, FZKA 6329, Forschungszentrum Karlsruhe
- Völkner, S. 2003 „Zum Einfluss räumlich begrenzter Diskontinuitäten auf die zeitabhängige Feuchteverteilung in Außenwänden“, Dissertation, Ruhr-Universität Bochum
- Technische Universität Braunschweig, Sonderforschungsbereich 477, „Sicherstellung der Nutzungsfähigkeit von Bauwerken mit Hilfe innovativer Bauwerksüberwachung“, 2000
- Johannes Weber, Wolfram Köhler: Zerstörungsfreie Untersuchungsmethoden an Steindenkmälern
- Kaatze, U. 2007 „Aspekte elektromagnetischer Aquametrie ionisch leitender Materialien“ in Technisches Messen, Ausgabe 5/2007, 261-267
Siehe auch Bearbeiten
Instrumentelle Analytik Bearbeiten

Die Instrumentelle Analytik (IA) ist der Bereich der Analytischen Chemie, der die Analyse und Identifikation von unbekannten Stoffen („Probe“) oder deren molekularen Strukturen mittels moderner Analysegeräte vornimmt. Hierzu wird eine Probe in der Regel vorbereitet (z. B. durch Extrahieren und Eluieren), in das Gerät gegeben und bestrahlt oder zerlegt, um in einem Detektor bestimmte Messwerte zu erfassen. Diese lassen auf die Art, Konzentration oder Menge der unbekannten Substanz schließen – oft bis in Bereiche winziger Mengen hinein.
Entwicklung und Merkmale Bearbeiten
.jpg)
Die instrumentelle Analytik beruht auf hochpräzisen Messprinzipien, die in den letzten 40 Jahren neu entwickelt worden sind, um explosionsartig gestiegenen Anforderungen aus Forschung, Umweltanalytik und Produktkontrolle entgegen treten zu können. Auch das Handling der Geräte (Labortechnik) wurde in diesem Zeitraum weiterentwickelt. Im Vergleich zu klassisch-nasschemischen Methoden sind es Merkmale instrumenteller Analyseverfahren, dass die Messungen mit Hilfe hochmoderner Analysegeräte schnell, preiswert, richtig, präzise und mit kleinsten Mengen durchgeführt werden können. Viele Stoffe (Prüfsubstanzen) sind so bis in den für Laien kaum vorstellbaren Bereich von wenigen Pikogramm hinein messbar geworden.
Zu einer genauen Beschreibung der jeweiligen instrumentellen Analysemethode (zum Beispiel bei der Untersuchung eines Fertiggerichtes auf eines Schädlingsbekämpfungsmittels im Gemüse) gehören neben der Prüfeinrichtung (das Labor) und dem Prüfsystem (hier: das Fertiggericht) die Angabe des Prüfgerätes (z. B. Gaschromatograph, GC), der Prüfsubstanz (das Pestizid) und der Probe (Marke des Fertiggerichtes „Zigeunerschnitzel mit Kartoffelpürree“, Herstellung des Extraktes aus z. B. 10 g Paprika mit Hilfe eines bestimmten Extraktionsmittels wie z. B. 50 ml Dichlormethan).
Die Qualität einer Analysemethode wird beschrieben, indem neben der Dokumentation der Probenvorbereitung (z. B. durch Elution oder Extraktion) auch die einer Methodenvalidierung erfolgt, also der Nachweis und die Dokumentation der Zuverlässigkeit einer Analysemethode). Hierzu wird die Richtigkeit, die Genauigkeit und die Präzision der Methode untersucht und im Falle der Genauigkeit mit Hilfe der Gauß-Funktion auch berechnet (z. B. bei der Messwertwiedergabe in „Peak“-Form). Die Kalibrierung stellt einen Zusammenhang zwischen Messgröße und Analyseergebnis her, die Zuverlässigkeit des Ergebnisses wird über den arithmetischen Mittelwert („Durchschnitt“), die Standardabweichung und die Wiederfindungsrate beschrieben.
Einteilung Bearbeiten
Unter den Oberbegriff instrumentelle Analytik fallen vier Gruppen von Verfahrensweisen: optische, spektroskopische, chromatographische und elektroanalytische Methoden.
Optische Analysemethoden Bearbeiten


→Einführender Artikel: Optik
Optische Methoden und Geräte sind solche, bei denen die Probe mit Licht bzw. lichtähnlicher elektromagnetischer Strahlung beleuchtet wird. Ein Teil dieser Strahlung wird dabei gebeugt oder in einem bestimmten Winkel reflektiert. Dieser kann dann in einem Refraktometer gemessen werden (Abb. links).
Zu diesen Methoden gehören neben der Refraktometrie (Messung der Brechzahl) auch die Polarimetrie (Messung der optischen Aktivität/des Drehwertes) und die Fotometrie (Messung der Lichtabsorption in Form der Extinktion oder Transmission bei einer bestimmten Wellenlänge). Die Polarimetrie z. B. ist eine Methode, bei der eine Probelösung mit linear polarisiertem Licht einer bestimmten Wellenlänge durchleuchtet wird. Bestimmte Substanzen haben die Fähigkeit, die Schwingungsebene des linear polarisierten Lichtes zu drehen (spezifische Drehung, optische Aktivität). Diese spezifische Drehung wird in der Pharmazie und Chemie oft zur Identifizierung und Reinheitskontrolle von chiralen Stoffen eingesetzt. Besondere Bedeutung besitzt die Angabe der spezifische Drehung für Naturstoffe, wie beispielsweise Aminosäuren, Terpene und Zucker, da die Mehrzahl dieser Stoffe optisch aktiv ist.
Spektroskopische Methoden Bearbeiten


Bei spektroskopischen Methoden und Geräten tritt die Probe in Wechselwirkung mit elektromagnetischer Strahlung (Absorption und Emission, vgl. Spektroskopie, Spektralanalyse). Die verschiedenen instrumentellen Analyseverfahren der Spektroskopie werden unterteilt nach:
- dem Wellenlängenbereich der eingesetzten Strahlung (z. B. Röntgen-, UV-, VIS_ und IR-Spektroskopie, vgl. Abbildung),
- der Energieform (z. B. kernmagnetische Resonanz / NMR, Elektronenspin- oder Schwingungs-Spektroskopie),
- der Untersuchungstechnik (Absorptions- oder Emissionsspektroskopie, z. B. AAS/AES).
Die Atomspektroskopie im Atomabsorptionsspektrometrometer (Abkürzung: AAS) ist allgemein eine Messung der Wellenzahl, Wellenlänge oder Frequenz absorbierter Strahlung zur Bestimmung atomarer Energieniveaus), die Atomemissionsspektroskopie im Atomemissionsspektrometer/Flammenphotometer (AES) misst abgestrahlte Energie, die IR-Spektroskopie (im Infrarotspektrometer IR) und die UV/VIS-Spektroskopie (im UV/VIS-Spektrometer) messen im infraroten, sichtbaren und ultravioletten Bereich des elrektromagnetischen Spektrums.
Eine Fluoreszenzspektroskopie misst das von einer fluoreszierenden Probe abgestrahlte Licht im Fluoreszenzspektrometer, während bei einer Röntgenfluoreszenzanalyse (RFA) Röntgenstrahlung zum Einsatz kommt. Die Kernspinresonanz (NMR) wird im Kernresonanzspektrometer gemessen, die Elektronenspinresonanz im Elektronenspinresonanzspektrometrie (ESR oder EPR).
Das Massenspektrometer (MS) hingegen ist ein Gerät, das nicht spektroskopisch arbeitet (keine Verwendung elektromagnetischer Strahlung), sondern die Moleküle der Probesubstanz in Fragmente zerlegt, die dann von einem Magnetfeld erfasst und rechnerisch nach Massenzahlen sortiert werden.
Chromatographische Methoden Bearbeiten


Chromatographische Methoden und Geräte verfolgen das Ziel, die Probe (ein Stoffgemisch) unter bestimmten Bedingungen in eine Bewegung zu versetzen, so dass die Komponenten auf Grund unterschiedlicher Fließ- oder Wanderungsgeschwindigkeit aufgeteilt und identifiziert werden können (Gas-, Hochdruck-Flüssigkeits-Chromatographie u. ähnl., Trennung mittels Adsorption und Desorption sowie unterschiedlicher Retentionszeiten der Einzelkomponenten). Es handelt sich hier also um hochpräzise Mikroverfahren zur Stofftrennung (im Vergleich zu den klassischen Makro-Stofftrennverfahren der Destillation, Sublimation, Extraktion, Umkristallisation, Umfällung und Filtration).
Wichtige Analysegeräte zur Chromatographie sind Gaschromatographen (GC, oft zur Bestimmung von Peakflächen und Retentionsfaktoren bzw. -zeiten von bestimmten Substanzen), HPLC-Geräte (zur Hochleistungsflüssigkeitschromatographie), Ionenchromatographen (zur Ionenchromatographie/Elektrophorese IC/EP) sowie Elektrophoreseapparaturen zur Gelelektrophorese, speziell 2D-Gelelektrophoresen und IEF-Gelelektrophoresen (IEF=Isoelektrische Fokussierung.
Elektroanalytische und weitere instrumentelle Analysemethoden Bearbeiten

Ferner existieren elektroanalytische und auch weitere physikalische Methoden und Geräte, bei denen die Probe z. B. elektrisch aufgeladen, dem elektrischen Strom ausgesetzt (Elektrolyse, Potentiometrie oder Konduktometrie u. ähnl.) oder in Molekülbruchstücke (Fragmente) zertrümmert und aufgetrennt wird (Massenspektrometrie MS). Die neun bedeutsamsten Verfahren dieser Gruppe sind:
- Elektrogravimetrie zur Konzentrationsbestimmung über die abgeschiedene Elektrolytmasse
- Potentiometrie (Voltametrie) zur Messung des elektrochemischen Potenzials bzw. der Spannung
- Polarographie/Voltammetrie Aufzeichnung von Strom-Spannungs-Kurven zur quantitativen und qualitativen Analyse
- Amperometrie Registrierung des Elektrolysestroms bei konstantem Potential
- Coulometrie Registrierung der elektrischen Ladung, die bei einer Elektrolyse übertragen wird
- Konduktometrie, z. B. als automatisierte Leitfähigkeitstitration
- Osmometrie zur Messung des osmotischen Druckes
- Viskosimeter zur Messung der Viskosität
- Massenspektrometrie zur Ermittlung der molaren Masse eines Moleküls bzw. seiner Schlüsselbruchstücke
Nasschemische bzw. klassische Analysemethoden wie die Volumetrie (Maßanalyse) und Gravimetrie (Fällungsanalyse) werden nur dann zu den instrumentellen Analysemethoden gerechnet, wenn elektronische Messinstrumente dabei eingesetzt werden. Bei der Maßanalyse wäre das die instrumentelle Indikation des Äquivalenzpunktes beispielsweise mittels Potentiometrie oder Konduktometrie. Eine instrumentelle Variante der Gravimetrie ist die Thermogravimetrie.
Gekoppelte Methoden Bearbeiten
In jüngere Zeit hat die direkte Kopplung unterschiedlicher Analysemethoden stark an Bedeutung gewonnen. Ein klassisches Beispiel ist die sog. GC-MS, bei der ein Massenspektrometer (MS) mit einem Gaschromatographen (GC) verbunden ist (GC-MS). Durch den Chromatographen wird eine Auftrennung eines oft komplexen Substanzgemischs erzielt, während das mit dem Gasfluss des Chromatographen kontinuierlich gespeiste Massenspektrometer eine Identifizierung der einzelnen Probenkomponenten ermöglicht. Ähnliche Kombinationen sind Kopplungen der High Performance Liquid Chromatography (HPLC) und Massenspektrometrie oder von HPLC und NMR.
Literatur Bearbeiten
- Heinz Hug: Instrumentelle Analytik – Theorie und Praxis. Haan-Gruitebn 2010, ISBN 978-3-8085-7211-5.
- Wolfgang Gottwald: Instrumentell-analytisches Praktikum. Weinheim/New York/Basel/Cambridge/Tokyo 1996, ISBN 3-527-28755-8.
- Wolf R. Less, Stefan Eckhardt u.a.: Die handlungsorientierte Ausbildung für Laborberufe. Band 2: Wahlqualifikationen. Würzburg 2006, ISBN 978-3-8343-3021-5, S. 87–378.
- Douglas A. Skoog, James J. Leary,: Instrumentelle Analytik : Grundlagen - Geräte – Anwendungen. Berlin 1996, ISBN 978-3-540-60450-1.
- Michael Wächter: Stoffe, Teilchen, Reaktionen – Basiswissen Chemie für Chemieberufe. Hamburg 2000, ISBN 3-582-01235-2, S. 177–202.
Spektroskopie Bearbeiten

Spektroskopie ist eine Gruppe von Methoden, das Energiespektrum einer Probe zu untersuchen, indem Strahlung nach ihrer Energie zerlegt wird. Zur visuellen Betrachtung optischer Spektren dienten Spektroskope, aufzeichnende Geräte heißen Spektrometer. Letztere arbeiten auch in anderen Bereichen des elektromagnetischen Spektrums sowie mit Teilchen wie Elektronen oder Ionen. Dabei kann die Anregung der Probe mit einer Strahlungsart erfolgen und dann eine andere Ausstrahlung der Probe untersucht werden.
Bereits 1814 entdeckte Joseph von Fraunhofer dunkle Linien im Spektrum der Sonne, die Fraunhofer-Linien, ohne allerdings ihren Ursprung erklären zu können. 1859 fanden Gustav Robert Kirchhoff und Robert Wilhelm Bunsen, dass verschiedene chemische Elemente die Flamme eines Gasbrenners auf charakteristische Weise färben. Diese Art von Anwendung zielt auf die Zusammensetzung der Probe. Wenn dabei quantitative Messungen erfolgen, oft von Konzentrationen, aber auch anderer physikalischer Größen wie Druck oder elektrischer oder magnetischer Felder, so spricht man von Spektrometrie. Dazu zählen im weiteren Sinne auch Methoden, bei denen nicht nach der Energie aufgelöst wird, sondern etwa nach der Masse von Teilchen, siehe Massenspektrometrie.
Oft geht es aber gerade um das Energiespektrum selbst. So gaben spektroskopische Beobachtungen entscheidende Impulse für die Entwicklung der Quantenmechanik und geben Hinweise auf die chemische Struktur unbekannter Substanzen. Die Präzision, mit der manche Spektrallinien gemessen werden können, erlaubt die Bestimmung von Naturkonstanten, den Test von Hypothesen über Naturgesetze und die Definition der Basiseinheiten Meter und Sekunde.
Physikalische Grundlagen Bearbeiten

Ein Spektrum im Sinne dieses Artikels ist die Auftragung einer Intensität über einer Energieskala (Frequenz, Wellenzahl, Wellenlänge). Der Zusammenhang zwischen der Frequenz einer elektromagnetischen Welle und der Energie der Lichtquanten ist dabei gegeben durch



mit der Planck-Konstanten .

Grundlage zum Verständnis von Spektren (ohne auf das eigentlich formal korrekte Orbitalmodell zurückzugreifen) ist das Bohrsche Atommodell. Mit diesem kann man Absorption und Emission von Photonen durch Übergänge zwischen verschiedenen Energieniveaus eines Atoms erklären. Die absorbierte bzw. emittierte Energie ist dabei durch das anfängliche Energieniveau (in der Quantenmechanik werden diese Energieniveaus als Zustand bezeichnet) und dem End-Energieniveau festgelegt.



Dabei gilt:

Ist , die Differenz also positiv, so handelt es sich in diesem Beispiel um Emission, bei negativen Vorzeichen, also dann um Absorption.


Strukturen im Spektrum geben Hinweise darauf, welche Energiebeträge eine Substanz aufnehmen (absorbieren) oder abgeben (emittieren) kann. Diese Beträge entsprechen Energiedifferenzen quantenmechanischer Zustände der Probe. Intensitäten im Spektrum hängen insbesondere ab von Konzentrationen, von Auswahlregeln und Besetzungszahlen.
Klassische Spektroskopie Bearbeiten
Die Untersuchung der Lichtemission bzw. -absorption von Molekülen und Atomen mit Hilfe von Gitter- und Prismenspektrometern sind die ältesten spektroskopischen Verfahren. Sie werden daher auch als klassische Spektroskopie bezeichnet. Viele der grundlegenden Untersuchungen über den Aufbau des Atoms wurden erst durch die Entwicklung und Anwendung hochauflösender Gitter- und Prismenspektrometer möglich.
Spektroskopiearten Bearbeiten
Einteilung Bearbeiten
Die Einteilung der zahlreichen spektroskopischen Methoden und Verfahren ist vielfältig und in der Literatur nicht immer einheitlich. Allgemein unterscheidet man zunächst zwischen Methoden der Atom- und der Molekülspektroskopie. Die Atomspektroskopie umfasst spektroskopische Verfahren, die auf Emissions-, Absorptions- oder Fluoreszenzvorgängen bei Atomen zurückgehen und zur Bestimmung von chemischen Elementen eingesetzt werden. Die beobachteten Spektren sind im Allgemeinen Linienspektren. Die molekülspektroskopischen Verfahren basieren hingegen auf der Anregung und Auswertung von Rotations-, Schwingungs- und Elektronenzuständen in Molekülen. Durch die Überlagerung von Einzelzuständen werden dabei keine Linienspektren sondern sogenannte Bandenspektren beobachtet.
Neben dieser grundlegenden Einteilung, nach der Art der untersuchten Zuständen, gibt es zahlreiche andere Unterteilungen, beispielsweise nach der Anregungsenergie der elektrischen Strahlung (z. B. Mikrowellenspektroskopie, Röntgenspektroskopie), des Aggregatzustandes (z. B. Festkörperspektroskopie) oder der Art der Anregung (z. B. Elektronenspektroskopie, Laserspektroskopie).
| EM-Strahlung | Wellenlänge | Frequenzbereich | Wellenzahl | Energiebereich | untersuchte Eigenschaft | Spektroskopische Methode |
|---|---|---|---|---|---|---|
| Radiowellen | 100 m…1 m | 3·106…300·106 Hz | 10−4…0,01 cm−1 | 10−6…10−4 kJ/mol | Änderung des Kernspinzustandes | Kernresonanzspektroskopie (NMR, auch Hochfrequenzspektroskopie) |
| Mikrowellen | 1 m…1 cm | 300·106…30·109 Hz | 0,01…1 cm−1 | 10−4…0,01 kJ/mol | Änderung des Elektronenspinzustandes oder Hyperfeinzustandes | Elektronenspinresonanz (ESR/EPR), Ramsey-Spektroskopie (Atomuhren) |
| Mikrowellen | 10 cm…1 mm | 30·108…3·1011 Hz | 0.1…10 cm−1 | 0,001…0,1 kJ/mol | Änderung des Rotationszustandes | Mikrowellenspektroskopie |
| Terahertzstrahlung | 1 mm…100 µm | 0,3·1012…30·1012 Hz | 10…100 cm−1 | 0,1…1 kJ/mol | Änderung des Schwingungszustandes | Submillimeterwellenspektroskopie |
| Infrarotstrahlung | 1 mm…780 nm | 3·1011…3,8·1014 Hz | 10…12,84 cm−1 | 0,12…153 kJ/mol | Änderung des Schwingungszustandes | Schwingungsspektroskopie; (Infrarotspektroskopie (IR), Reflexionsspektroskopie und Ramanspektroskopie, Ultrakurzzeit-Spektroskopie) |
| sichtbares Licht; UV-Strahlung | 1 µm…10 nm | 3·1014…3·1016 Hz | 104…106 cm−1 | 100…104 kJ/mol | Änderung des Zustandes der äußeren Elektronen | UV/VIS-Spektroskopie (UV/Vis), Reflexionsspektroskopie, Fotoleitungsspektroskopie, Fluoreszenzspektroskopie; Ultrakurzzeit-Spektroskopie; Atomspektroskopie; Vergleich mit Frequenzkamm |
| Röntgenstrahlung | 10 nm…100 pm | 3·1016…3·1018 Hz | 106…108 cm−1 | 104…106 kJ/mol | Änderung des Zustandes der Rumpfelektronen | Röntgenspektroskopie (XRS); Elektronenspektroskopie; Auger-Elektronen-Spektroskopie (AES); Mößbauer-Spektroskopie |
| Gammastrahlung | 100 pm…1 pm | 3·1018…3·1020 Hz | 108…1010 cm−1 | 106…108 kJ/mol | Änderung des Kernzustandes (Anordnung der Nukleonen) | Gammaspektroskopie |
Liste mit Spektroskopiearten und -methoden in der Analytik Bearbeiten
- Atomspektroskopie – Messungen der Eigenschaften einzelner Atome, vor allem ihrer Elektronen-Energieniveaus
- Atomabsorptionsspektroskopie (AAS/OAS)
- Atomemissionsspektroskopie (AES/OES)
- Induktiv gekoppeltes Plasma (ICP-AES)
- Mikrowellen-Plasmafackel-AES (MPT-AES)
- Atomfluoreszenzspektroskopie (AFS)
- Gammaspektroskopie
- Mößbauer-Spektroskopie (beruhend auf dem Mößbauer-Effekt)
- Elektronenspektroskopie
- Röntgenspektroskopie (XRS)
- Glimmentladungsspektroskopie (GDOES)
- Molekülspektroskopie – Messungen der Eigenschaften einzelner Moleküle, vor allem der Valenzelektronen-Energieniveaus und der Molekülschwingungen und -rotationen
- Frequenzmodulationsspektroskopie
- Fluoreszenzspektroskopie
- Schwingungsspektroskopie
- Kernresonanzspektroskopie (NMR, auch Hochfrequenzspektroskopie)
- Elektronenspinresonanz (ESR/EPR)
- Elektron-Kern-Doppelresonanz (ENDOR)
- Mikrowellenspektroskopie
- UV/VIS-Spektroskopie (UV/Vis)
- Festkörperspektroskopie – Messungen der Eigenschaften ganzer Festkörper (wie Kristalle), vor allem deren Bandstrukturdetails
- Impedanzspektroskopie (Dielektrische Spektroskopie)
- Laserspektroskopie
- Cavity-ring-down-Spektroskopie (CRDS, auch CRLAS)
- Laserinduzierte Fluoreszenz (LIF)
- Ultrakurzzeit-Spektroskopie – Messungen der Details von schnellen Vorgängen, vor allem chemischer Reaktionen
Spektroskopie in der Astronomie Bearbeiten

Das Element Helium wurde – lange vor dem Nachweis auf der Erde – zunächst durch spektroskopische Untersuchungen des Sonnenlichtes erkannt. Eine Spektrallinie im Sonnenspektrum konnte keiner laborchemischen Substanz zugeordnet werden, so dass auf der Sonne ein unbekanntes Element existieren musste.
Weitere klassische Erfolge der astronomischen Spektralanalyse sind
- der Nachweis des Doppler-Effektes an Sternen (siehe auch Radialgeschwindigkeit)
- und (um 1920) an Galaxien (siehe Rotverschiebung),
- von Magnetfeldern auf Sonne und hellen Sternen (Zeeman-Effekt)
- und vor allem die Feststellung von Stern-Temperaturen und der Spektralklassen (siehe auch Hertzsprung-Russell-Diagramm und Sternentwicklung).
Die zugehörigen Messinstrumente („Spektralapparate“) der Astrospektroskopie sind:
- das Spektroskop und das Spektrometer (beides visuell)
- der Spektrograf (fotografisch bzw. mit Sensoren)
- der Monochromator und das Interferenz-Spektrometer
- der Frequenzkamm[1]
Siehe auch Bearbeiten
Literatur Bearbeiten
Allgemeine Lehrbücher Bearbeiten
- Ludwig Bergmann, Clemens Schaefer: Lehrbuch der Experimentalphysik: Band 1 bis 8. De Gruyter.
- Peter M. Skrabal: Spektroskopie, Eine methodenübergreifende Darstellung vom UV- bis zum NMR-Bereich. vdf Hochschulverlag AG, Zürich 2009, ISBN 978-3-8252-8355-1.
- Physikalische Chemie#Literatur
Spezielle Werke Bearbeiten
deutsch
- Wolfgang Demtröder: Molekülphysik: Theoretische Grundlagen und experimentelle Methoden. 1. Auflage. Oldenbourg, 2003, ISBN 3-486-24974-6.
- Wolfgang Demtröder: Laserspektroskopie: Grundlagen und Techniken. 5. Auflage. Springer, Berlin 2007, ISBN 978-3-540-33792-8.
- Hermann Haken, Hans Christoph Wolf: Atom- und Quantenphysik. 8. Auflage. Springer, Berlin 2003, ISBN 3-540-02621-5.
- Hermann Haken, Hans Christoph Wolf: Molekülphysik und Quantenchemie. 5. Auflage. Springer, Berlin 2006, ISBN 3-540-30314-6.
englisch
- Peter W. Atkins, Ronald Friedman: Molecular Quantum Mechanics. 4. Auflage. Oxford University Press, Oxford 2004, ISBN 0-19-927498-3.
- Peter F. Bernath: Spectra of Atoms and Molecules. 2. Auflage. Oxford University Press, Oxford 2005, ISBN 0-19-517759-2.
- Wolfgang Demtröder: Atoms, Molecules and Photons. Springer, Berlin 2005, ISBN 3-540-20631-0.
- Jack D. Graybeal: Molecular Spectroscopy. McGraw-Hill Education, New York NY u. a. 1988, ISBN 0-07-024391-3.
- J. Michael Hollas: Modern Spectroscopy. 4. Auflage. John Wiley & Sons, Chichester 2003, ISBN 0-470-84416-7.
- E. Bright Wilson Jr., J. C. Decius, Paul C. Cross: Molecular Vibrations – The Theory of Infrared and Raman Vibrational Spectra. Dover Publications, New York NY 1980, ISBN 0-486-63941-X.
- Gordon G. Hammes: Spectroscopy for the biological sciences. Wiley-Interscience, Hoboken NJ 2005, ISBN 0-471-71344-9.
Weblinks Bearbeiten
- Commons: Bin im Garten/Chemie/Brückenkurs – Sammlung von Bildern, Videos und Audiodateien
- Grundlagenforschung im Bereich der Spektroskopie
- Maßgeschneiderte Medikamente per Laser Fluoreszenz-Korrelations-Spektroskopie (FCS) Universität Bonn
- The Science of Spectroscopy – hervorragende Seite zum Thema nach dem Wiki-Prinzip (engl.)
Massenspektrometrie Bearbeiten
Als Massenspektrometrie werden Verfahren zum Messen der Masse von Atomen oder Molekülen bezeichnet. Die Massenspektrometrie zählt nicht zu den Methoden der Spektroskopie, da es nicht um Spektren von elektromagnetischer Strahlung geht.
Die zu untersuchende Substanz, der Analyt, wird in die Gasphase überführt und ionisiert. Die Ionen werden durch ein elektrisches Feld beschleunigt und dem Analysator zugeführt, der sie nach dem Masse-zu-Ladung-Verhältnis m/q "sortiert", beispielsweise (im Sektorfeld-Massenspektrometer, siehe unten) räumlich in Teilstrahlen auftrennt. Die Moleküle können dabei fragmentiert werden.[2][3]
Massenspektrometrie findet in vielen Bereichen Anwendung. Eingesetzt wird sie unter anderem bei der Charakterisierung von chemischen Verbindungen, in der medizinischen Chemie zur Identifizierung von Substanzen in Körperflüssigkeiten oder Organen, in kriminaltechnischen Untersuchungen, Dopingkontrollen, der militärischen Analytik von chemischen Kampfstoffen und in der Pharmakokinetik. Es gibt sehr unterschiedliche Techniken, die sich je nach Aufwand, Anwendung und Genauigkeit unterscheiden.
Vorteilhaft ist in vielen Bereichen, dass die Datenmenge recht gering ist und damit eine Kopplung mit Datenbanken von Massenspektren leicht möglich ist. Es ist auch verhältnismäßig leicht ein Massenspektrometer mit einer HPLC-Anlage oder einem Gaschromatographen zu koppeln und so die verschiedenen Massenspektren der im Analyten enthaltenden Verbindungen zu erhalten.
Geschichte Bearbeiten
Die Massenspektrometrie basiert auf einer Hypothese, die vom britischen Chemiker William Prout im frühen 19. Jahrhundert aufgestellt wurde und besagt, dass es eine Eigenschaft eines Atoms ist, eine bestimmte Masse zu haben – das Atomgewicht. Er hatte beobachtet, dass sich Atome von ihrer Masse her als ganzzahliges Vielfaches der Masse von Wasserstoff-Atoms verhalten. [4] [5] Spätere und genauere Messungen von Jöns Jakob Berzelius (1828) und Edward Turner (1832) schienen jedoch diese Hypothese zu widerlegen, denn es wurde z. B. für das Chlor-Atom eine Masse bestimmt, die das 35,45-fache der Wasserstoffmasse beträgt.

In der Mitte des 19. Jahrhunderts beobachtete Julius Plücker den Einfluss von magnetischen Feldern auf das Leuchten von Gasentladungsröhren.
Eugen Goldstein und Wilhelm Wien publizierten in den Jahren 1886 und 1898 die sogenannten Kanalstrahlen und ihre Ablenkung durch Felder. Sie hatten die weitreichenden Konsequenzen ihrer Entdeckungen jedoch 1886 noch nicht erkannt. [6] [7] [8] [9] [10] [11]

Später, im Jahr 1897, publizierte J. J. Thomson verschiedene Experimente, in denen er in Vakuumröhren Kathodenstrahlen von verschiedenen Kathodenmetallen mit elektromagnetischen Feldern ablenkte, und stellte korrekte Gleichungen zum Zusammenhang zwischen Masse, Geschwindigkeit und Bahnradius auf. 1913 publizierte er eine Methode, um mit Hilfe eines Massenspektroskops Fotoplatten zu belichten und so qualitative und quantitative Untersuchungen an den in einer Röhre enthaltenen Gasen durchzuführen.
Ein Schüler von Thomson, der britische Chemiker und Physiker Francis William Aston, baute 1919 das erste funktionierende Massenspektrometer. Mit dieser neuen Technik konnte er die Isotope von Chlor (35Cl und 37Cl), von Brom (79Br und 81Br) und von Krypton (78Kr, 80Kr, 82Kr, 83Kr,84Kr und 86Kr) beobachten. Aston wurden schließlich im Jahr 1922 mit dem Nobelpreis für Chemie für seine Untersuchungen der Isotope geehrt. Durch die Verwendung der Technik des Elektrofokusierens konnte er nicht weniger als 212 der damals bekannten 287 Isotope beobachten.
Im Jahr 1918 wurde von Arthur Jeffrey Dempster das erste moderne Massenspektrometer entworfen und gebaut, welches 100 fach genauer arbeitete als alle vorherigen Entwicklungen und legte den Grundstein für das Design heutiger Massenspektrometer. Aufgrund dieser Entwicklung hat er im Jahr 1935 das das Uranisotop mit der Masse 235 identifizieren können.
Während des Manhattan-Projekts wurden riesige Isotopenanreicherungsanlagen auf Grundlage der Massenspektrometrie gebaut.

In den 1950er Jahren wurde von Roland Gohlke und Fred McLafferty das erste Mal ein Massenspektrometer als Detektor für eine Chromatographie-Methode eingesetzt. Beide koppelten einen Gaschromatographen mit einem Massenspektrometer. Durch diese Methode konnten das erste Mal Substanzgemische in einer Anlage getrennt und identifiziert werden.[12][13] Für die Anwendung in einer gaschromatographischen müssen die entsprechenden Verbindungen jedoch im Vakuum flüchtig und dabei unzersetzlich verdampfbar sein.
Im Jahr 1974 entwickelten Alan G. Marshall und Melvin B. Comisarow von der University of British Columbia inspieriert von den Fourier Transform Nuclear Magnetic Resonance (FT-NMR) und ICR Methoden ein Fouriertransform-Massenspektrometer.[14]
Die bisherigen Methoden zur Erzeugung der benötigten Ionen waren sehr aggressiv und führten zu vielen Bruchstücken (Fragmente) beim vermessen organischer Verbindungen. Daher setzte ab den 1960 Jahren die Entwicklung immer schonenderer Ionisationsmethoden ein. Mitte der 1960er Jahre wurde von Munson und Field die Chemische Ionisation (CI) veröffentlicht.[15] Im Jahr 1969 hat Beckey die Feld Desorbtion (FD) publiziert.[16]
Später erfolgten dann die weitere Entwicklung einer Vielzahl an Ionisationsmethoden für die unterschiedlichsten Zwecke wie zum Beispiel Chemische Ionisation bei Atmosphärendruck (APCI), Matrix-unterstützte Laser-Desorption/Ionisation (MALDI), Elektrospray (ESI).
Parameter eines Massenspektrometers Bearbeiten
Ein Massenspektrometer wird im Wesentlichen durch zwei Parameter charakterisiert: Die Massenauflösung und die Massengenauigkeit.
Die Massenauflösung bezeichnet den minimalen Massenunterschied dm, den zwei Ionen haben müssen, damit sie noch aufgelöst werden können. Die Auflösung eines Massenspektrometers wird in der Einheit Thomson (Th) angegeben, wobei aber trotzdem öfter nur das Auflösungsvermögen R angegeben wird. Dieses ist als Verhältnis einer Masse zum Massenunterschied der nächsten noch getrennt erscheinenden Masse ( ) definiert. Zum Beispiel würde man bei einem Auflösungsvermögen von 4000 die Peaks bei 4000 Th und 4001 Th noch getrennt sehen, aber ebenso die Peaks bei 2000 Th und 2000,5 Th da 2000/(2000,5 − 2000) = 4000. In der Praxis werden die beiden Begriffe Auflösung und Auflösungsvermögen leider oft nicht exakt auseinander gehalten.

Es gibt drei verschiedene Definitionen der Auflösung:
- Bei der 10-%-Methode definiert man dm als die Massenabweichung, bei der die Intensität eines Peaks auf 10 % des Maximums absinkt.
- Bei der 50-%-Methode definiert man dm als die Massenabweichung, bei der die Intensität eines Peaks auf 50 % des Maximums absinkt.
- Bei der Halbwertsbreitenmethode (FWHM (full width at half maximum height)) definiert man dm als die volle Peakbreite bei der halben Peakhöhe.
Die Massengenauigkeit gibt an, wie genau die Masse des Teilchens bestimmt werden kann. Diese Angabe erfolgt oft in parts per million (ppm), d. h., ein Molekül mit der nominellen Masse 500 kann bei einer Genauigkeit von 1 ppm auf 0,0005 u genau bestimmt werden.
Aufbau eines Massenspektrometers Bearbeiten

Ein Massenspektrometer (MS) besteht aus einer Ionenquelle, einem Analysator und einem Detektor. Jedes dieser Bauteile existiert in verschiedenen Bauformen und Funktionsprinzipien, die prinzipiell frei kombinierbar sind, obschon bevorzugte Kombinationen existieren. Diese werden im Folgenden beschrieben.
Ionenquelle Bearbeiten
In der Ionenquelle wird der Analyt ionisiert. Dies kann mit Hilfe verschiedener Methoden erfolgen. Die Wahl der Methode ist hauptsächlich abhängig von der Art der zu analysierenden Substanz und davon, wie schonend ionisiert werden soll. Die Ionen werden meistens mit einem elektrischen Feld aus der Ionenquelle extrahiert und in den Analysator übergeben. Die einzelnen Methoden werden im Abschnitt Ionisationsmethoden behandelt.
Analysator Bearbeiten
Im Analysator oder Massenselektor werden die Ionen nach ihrer Masse (genauer: Masse-zu-Ladung-Verhältnis, also m/q) getrennt. Es gibt mehrere sehr unterschiedliche Methoden, wie diese Massentrennung erfolgt. Abhängig von der Methode ist auch das Trennvermögen recht unterschiedlich. Die einzelnen Trennmethoden werden im Abschnitt Arten von Analysatoren behandelt.
Detektor Bearbeiten
Der Detektor dient zur Erfassung der zuvor separierten Ionen. Als Detektor eingesetzt werden können Photomultiplier, Sekundärelektronenvervielfacher (SEV), Faraday-Auffänger, Daly-Detektoren, Mikrokanalplatten( MCP) oder Channeltrons. Der SEV wird teilweise in Kombination mit einer Konversionsdynode verwendet, bei der die Ionen aufgrund einer angelegten hohen Beschleunigungsspannung (bis zu 25 kV) auf eine Metalloberfläche prallen und der SEV dann die freiwerdenden Elektronen detektiert. In der Anfangszeit der Massenspektrometrie wurden auch Fotoplatten benutzt.
FT-ICR- und Orbitrap-Massenspektrometer messen Ströme (engl. image-currents), welche durch die sich bewegenden Ionenpakete in den Detektorplatten erzeugt werden. In diesem Fall werden die Ionen also nicht vom Detektor absorbiert und können deshalb mehrfach gemessen werden. Das trägt entscheidend zur hohen Messgenauigkeit dieser Instrumente bei.
Ionisationsmethoden Bearbeiten
Elektronenstoßmethoden Bearbeiten
- Elektronenstoßionisation (EI, auch Elektronenstoß, engl. electron impact)
- Die Elektronenstoßionisation ist die meistgenutzte Ionisationsmethode. [17] Dabei werden von einem Filament Elektronen mittels eines elektrischen Feldes auf eine kinetische Energie von 5 bis 200 eV (aus Stabilitätsgründen verwendet man oft 70 eV; außerdem ist der Ionisationsquerschnitt bei dieser Energie für die meisten Gase maximal) beschleunigt und durch eine Gaswolke der zu ionisierenden Moleküle geschickt. Beim Zusammenstoß der Elektronen mit den Molekülen wird ein weiteres Elektron herausgeschlagen, es entsteht ein Radikalkation. Dieses ist meistens instabil und zerfällt in kleinere Massenfragmente, von denen eines geladen bleibt. Die Größen und Häufigkeiten der Fragmente sind bei gegebener Beschleunigungsspannung der Elektronen für eine Substanz in Datenbanken katalogisiert und können zur Identifizierung herangezogen werden.
- Chemische Ionisation (CI)
- Bei der chemischen Ionisation wird ein Gas zugeführt, das durch Elektronenstoßionisation angeregt/ionisiert wird. Die aus dem Gas gebildeten Ionen reagieren dann mit dem Analyten und ionisieren ihn. Der Fragmentierungsgrad ist geringer als bei der Elektronenionisation.
- Sanfte Ionisation
- Bei der sanften Ionisation werden Gase mit geringer Ionisationsenergie von 10 bis 14 eV zur Ionisation der Probengase verwendet (z. B. Xenon, Argon oder Quecksilber). Dazu wird eine Ionenwolke des Ionisationsgases mittels Elektronenbeschusses erzeugt. Durch eine Auswahl über Vorfelder erreichen nur einfach ionisierte Atome des Ionisationngases das zu ionisierende Probengas. Eine Fragmentierung der Moleküle des Probengases wird dadurch nahezu ganz unterbunden. Da sich zudem unterschiedliche Gase mit gleicher Massenzahl von den Ionisierungsgasen verschieden ionisieren lassen, ist eine Differenzierung von Isomeren durch analytische Vergleiche möglich.
Feldionisationsmethoden Bearbeiten
- Feldionisation (FI)
- Bei der Feldionisation wird ein Gas in einem starken elektrischen Feld an zahlreichen Spitzen (Graphitdendriten) sehr schonend ionisiert.
- Felddesorption (FD)
- Bei der Felddesorption wird ein fester oder flüssiger Analyt, der den zahlreichen Graphitdendriten in Lösung zugeführt wird, in einem hohen elektrischen Feld wie bei FI sehr schonend ionisiert.
- Liquid Injection Field Desorption Ionization (LIFDI)
- Bei der LIFDI wird durch den Druckunterschied zwischen Normaldruck und Vakuum ein flüssiger Analyt über eine Kapillare direkt auf den Graphitdendriten gespritzt und dort in einem hohen elektronischen Feld wie bei FI sehr schonend ionisiert. Da die Zuführung des Analyt durch eine Kapillare erfolgt, ist diese Methode speziell für das Analysieren von luftempfindlichen Substanzen geeignet.
Teilchenbeschussmethoden Bearbeiten
Flüssigkeiten und Feststoffe können mit schnellen Atomen oder Ionen beschossen werden, worauf sich Ionen lösen. Kommen Atome zum Einsatz, heißt die Methode FAB (engl. Fast Atom Bombardment, Schneller Atombeschuss), bei Ionen SIMS (engl. secondary ion mass spectrometry, Sekundärionen-Massenspektrometrie). Neben den Sekundärionen werden auch ungeladene Teilchen (Sekundärneutralteilchen) erzeugt. Wenn diese zum Beispiel mit Laserlicht nachionisiert und dann analysiert werden, spricht man von Sekundär-Neutralteilchen-Massenspektrometrie (SNMS).
Sprühmethoden Bearbeiten
- Elektrospray-Ionisation
- Bei der Elektrospray-Ionisation (ESI) werden Lösungen geladener oder polarer Substanzen versprüht, ionisiert und die Tröpfchen dann getrocknet, so dass Ionen des Analyten zurückbleiben. Diese Methode ist besonders geeignet für größere Moleküle, wie beispielsweise Proteine.
- Atmospheric Pressure Chemical Ionization
- Die chemische Ionisation unter Atmosphärendruck (engl. Atmospheric Pressure Chemical Ionization, APCI) funktioniert ähnlich wie ESI, nur dass die Lösung des Analyten vor der Ionisation verdampft wird. Die Lösemittelmoleküle werden an einer spitzen Elektrode bei Atmosphärendruck ionisiert. Die Methode ist auch für weniger polare Analyten geeignet.
Photoionisationsmethoden Bearbeiten
- Photoionisation unter Atmosphärendruck
- Eine weitere Form der Ionisation unter Atmosphärendruck wird durch Photonen erreicht. Je nach Quelle der Photonen unterscheidet man zwischen Atmosphärendruck-Photoionisation (engl. Atmospheric Pressure Photo Ionization, APPI) mittels einer Entladungslampe oder Atmosphärendruck-Laserionisation (engl. Atmospheric Pressure Laser Ionization, APLI) mittels eines Lasers. Diese Ionisationsmethode wird hauptsächlich bei der Kopplung von Massenspektrometern mit LC-Systemen verwendet. Der Eluent wird zunächst verdampft und anschließend durch Photonen ionisiert. Dabei werden die Photonen senkrecht zum Molekülstrahl ausgesandt.
- Matrix-unterstützte Laser-Desorption/Ionisation
- Bei der Matrix-unterstützten Laser-Desorption/Ionisation (MALDI) wird der Analyt mit einem großen Überschuss einer Matrix genannten Substanz gemischt und cokristallisiert (Einbindung des Analyten in die kristalline Matrix). Die Matrix hat die Eigenschaft, beim Beschuss mit einem Laser bestimmter Wellenlänge Energie zu absorbieren (zum Beispiel Stickstofflaser 337 nm). Durch den Beschuss mit dem Laser wird der intakte Analyt verdampft, an den ein von der Matrix bereitgestellter Ladungsträger, z. B. ein Proton, gebunden ist.
- Ein-Photonen-Ionisation
- Bei der Ein-Photonen-Ionisation (engl. Single Photon Ionization, SPI) wird die Ionisationsenergie durch ein einzelnes Photon überwunden, dessen Wellenlänge im Vakuum-Ultraviolett-Bereich (VUV) liegt. Diese hochenergetischen Photonen können entweder als Synchrotronstrahlung oder mit Entladungslampen erzeugt werden.
- Resonanzverstärkte Mehrphotonenionisation
- Die resonanzverstärkte Mehrphotonenionisation (engl. Resonance Enhanced Multi Photon Ionization, REMPI) basiert auf der nahezu gleichzeitigen Absorption mehrerer ultravioletter Photonen, deren Gesamtenergie oberhalb der Ionisationsenergie des zu ionisierenden Moleküls liegt.
Weitere Methoden Bearbeiten
- Thermische Ionisation (TIMS, thermische Ionisationsmassenspektrometrie) wird in der Festkörpermassenspektrometrie eingesetzt. Dabei wird die Probe (Probenmenge je nach Stoff ng bis µg) zum Beispiel auf ein Wolframfilament aufgebracht. Durch das Filament wird Strom geschickt, wobei es sich erhitzt und die aufgebrachte Probe verdampft. Ein Teil der abgedampften Atome wird dabei ionisiert.
- Ein induktiv gekoppeltes Plasma (engl. inductively coupled plasma, ICP) bricht die meisten Verbindungen in ihre Elemente auf und es entstehen vorwiegend einfach positiv geladene Ionen. Diese Methode wird daher vor allem bei der Massenspektrometrie mit induktiv gekoppeltem Plasma in der anorganischen Elementanalytik und Spurenanalytik eingesetzt.
- Bei der Ionisation durch Glimmentladung (engl. glow discharge ionization, GDI) wird die Probe durch ein durch einen Lichtbogen erzeugtes Plasma ionisiert.
Arten von Analysatoren Bearbeiten
Massenspektrometer werden durch den jeweilig eingesetzten Analysator typisiert.
Sektorfeld-Massenspektrometer Bearbeiten

In Sektorfeld-Massenspektrometern werden die Ionen in elektrischen und magnetischen Feldern abgelenkt. Der Radius der Kreisbahnen, die sie in den Feldern durchlaufen, hängt von der Energie (im elektrischen Feld) und vom Impuls (im magnetischen Feld) der Ionen ab. In Kenntnis der Ladung, der Energie und des Impulses kann dann die Masse bestimmt werden. Sektorfeld-Massenspektrometer können so gebaut werden, dass Ionen mit leicht unterschiedlicher Geschwindigkeit auf einem Punkt im Detektor abgebildet werden (Geschwindigkeitsfokussierung). Auch Ionen, deren Flugbahn leicht geneigt ist, können auf einen Punkt abgebildet werden (Richtungsfokussierung). Massenspektrometer, die beides gleichzeitig können, nennt man doppelfokussierend. Die Fokussierung ist nötig, um bei hoher Auflösung noch eine akzeptable Intensität des Messsignals zu erhalten. Sektorfeld-Massenspektrometer erreichen Auflösungen von bis zu 100.000 und waren vor der Entwicklung der FT-Ionenfallen die Massenspektrometer mit der größten Auflösung. Heute werden sie nur noch selten verwendet, zum Beispiel in der Stabilisotopenmassenspektrometrie.

Quadrupol-Massenspektrometer Bearbeiten
Im Quadrupol-Massenspektrometer werden die erzeugten Ionen durch ein statisches, elektrisches Feld beschleunigt und durchfliegen zentral vier parallel liegende Stabelektroden, deren Schnittpunkte mit einer Ebene senkrecht zur Zylinderachse ein Quadrat bilden (Quadrupol). Im Wechselfeld zwischen den Quadrupol-Stäben findet eine m/q-Selektierung statt, so dass jeweils nur Teilchen mit einer definierten Masse das Feld durchlaufen können. Die Ionen treffen in einem Detektor mit Messverstärker auf, der den Ionenstrom misst und von der Software des angeschlossenen PCs zur Zählrate bzw. zum Partialdruck umgerechnet wird. Die Ionisationseinheiten, Quadrupole und Detektoren sind in unterschiedlichsten Varianten für unterschiedliche Anwendungen erhältlich. Es ist in teuren, hochauflösenden aber auch günstigen Varianten (als Restgasanalysator) zu erhalten und hat dadurch eine hohe Verbreitung im F&E-Bereich.


Massen-Selektierung im Quadrupol-Feld Bearbeiten
Die gegenüberliegenden Elektroden des Quadrupol befinden sich auf gleichem Potential und zwischen benachbarten Elektroden wird eine Spannung mit einem Gleich- und einem hochfrequenten Wechselspannungs-Anteil angelegt, d. h. . Die Bahn der Ionen im QMS wird durch die Differentialgleichungen von Mathieu beschrieben. Aus systematischen Untersuchungen dieser Differentialgleichungen ist bekannt, dass es gewisse stabile und instabile Bereiche gibt. Die Arbeitsgerade, d. h., die Gerade, auf der alle beobachtbaren Massen liegen, wird durch das Verhältnis bestimmt. Um ein möglichst gutes Auflösungsvermögen (in der Praxis R = 1.000 bis 4.000) zu erreichen, muss gelten. Der Schnitt aus Arbeitsgeraden und stabilem Bereich der Mathieuschen Differentialgleichungen ist dann sehr klein. Der Wert 0,1678 darf aber auf keinen Fall überschritten werden, ansonsten sind alle Ionen instabil. Instabile Ionen kollidieren während des Durchlaufes mit einem der vier Stäbe.



Über die Einstellung der Frequenz oder Spannungen lässt sich festlegen, welche Teilchen mit welchem Masse-zu-Ladungs-Verhältnis den Detektor über die zentrale Flugbahn erreichen. Die Bahn eines Teilchens mit richtigem m/e-Verhältnis ist sinusförmig mit gleichbleibenden Abständen zur Mittelbahn des Quadrupols. Alle anderen Ionen fliegen zwar auch im Sinustakt durch das Wechselfeld um diesen Sollbahnbereich, werden aber immer weit herausbeschleunigt, so dass sie irgendwann seitlich außerhalb des Quadrupols hinausschießen und den Einflussbereich des EM-Feldes verlassen.


Da die Ionisierung durch Elektronenstoß erfolgt, treten bei fast allen Atomen/Molekülen statt einem Peak bei der entsprechenden Masse des Atomes/Moleküles mehrere Peaks im Massenspektrum auf. Dies hat verschiedene Ursachen:
- Die Moleküle können bei der Kollision mit den Elektronen in verschiedene Bestandteile zerbrechen (Fragmentierung). So ruft z. B. Ethanol (C2H5OH) (Masse 46,07 g/mol) einen schwachen Peak bei 46 Th (C2H5OH) hervor, einen stärkeren Peak bei 45 Th (ein Wasserstoffatom fehlt) und den stärksten Peak (ca. 10-mal höher als die anderen Peaks) bei 31 Th (CH2OH). Weitere Peaks sind u. a. 17 Th (OH).
- Eine teilweise Rekombination oder Neubildung von Molekülen ist zwar sehr viel seltener. Es ist aber wegen der hohen Genauigkeit des Quadrupol-Massenspektrometers nicht auszuschließen, dass entsprechende Zählraten auftreten können.
- Die Bruchstücke der Moleküle bzw. Atome können zweifach oder sogar dreifach ionisiert werden. Da das Quadrupol-Massenspektrometer – wie andere Massenspektrometer – prinzipbedingt nur den m/q-Wert misst, ergeben sich folglich mehrere Peaks, z. B. Argon bei 40 Th und bei 20 Th.
- Bei hohen Konzentrationen von Edelgasen in einer Probe sind die sogenannten Cluster zu sehen, z. B. für Argon 40 ein Peak bei 80AMU.
Flugzeitmassenspektrometer (TOFMS) Bearbeiten
Im Flugzeitmassenspektrometer (TOFMS) wird ausgenutzt, dass die Ionen beim Eintritt in den Analysator alle die gleiche Energie haben und leichte Ionen deshalb schneller sind als schwere. Daher erreichen beim Flug durch einen feldfreien Raum leichte Ionen den Detektor eher als schwere Ionen. Der Flugzeitanalysator besteht somit nur aus einem Rohr unter Vakuum mit einem sehr schnellen Detektor am Ende. Die Auflösung beträgt bis zu R = 15.000 (10-%-Methode). In der Praxis haben sich Geräte mit Ionenspiegeln bzw. Reflektron bewährt, bei denen die Flugstrecke durch ein zusätzliches elektrisches Feld am Ende der ursprünglichen Flugrichtung verdoppelt wird. Zusätzlich erreicht man durch diese Technik eine weitere Fokussierung, die die Varianz in der Geschwindigkeit der Ionen aufgrund des Doppler-Effekts minimiert.
Ionenfallen-Massenspektrometer Bearbeiten

In Ionenfallen-Massenspektrometern werden die Ionen durch elektromagnetische Felder in einem definierten Bereich gehalten und können so analysiert und manipuliert werden. In Ionenfallen-Massenspektrometern ist eine mehrfache Wiederholung von Anregung und Massenselektion möglich, ohne dass ein weiteres Bauteil benötigt wird. Folgende Typen von Ionenfallen-Massenspektrometer existieren:
- Quadrupol-Ionenfalle
- Linear trap
- Fouriertransformations-Ionenzyklotronresonanz (FT-ICR)
- Orbitrap
MS/MS (auch Tandem-Massenspektrometrie) Bearbeiten
Um Fragmentierungen zu studieren oder auch um die Selektivität und Sensitivität (Nachweisgrenze) einer Quantifizierungsmethode entscheidend zu verbessern, koppelt man entweder mehrere Analysatoren hintereinander oder arbeitet in Ionenfallen. Zwischen zwei Analysatoren wird eine sogenannte Kollisionszelle eingebaut, um den Ionen durch Stöße mit einem Inertgas (meist Stickstoff oder Argon) Energie zuzuführen. Daraufhin zerfallen die Ionen sehr spezifisch zu anderen (leichteren) Ionen.
Viele Kombinationen der Analysatoren sind denkbar. Die gängigsten sind Triple-Quadrupol (QqQ), Doppel-Quadrupol (Qq), Tandem-TOF (TOF-TOF) und inzwischen auch TRAP-FTICR und TRAP-Orbitrap.
Am weitesten verbreitet sind sogenannte Triple-Quadrupol (QqQ, auch triple quads genannt). Dabei wird meist durch ESI ein Pseudomolekülion produziert, im ersten Analysatorquadrupol isoliert und dann im zweiten Quadrupol – der sogenannten Kollisionszelle oder Stoßkammer – angeregt.
In die Stoßkammer kann ein Stoßgas (meist Argon, Helium oder Stickstoff) eingespeist werden. Dabei wird der Druck so gewählt, dass im Mittel ein erzeugtes Ion maximal einmal mit einem Gasmolekül kollidiert. Diese Methode ermöglicht es, erzeugte Ionen weiter zu fragmentieren.
Der dritte Quadrupol gibt die Möglichkeit zu „scannen“, also alle Produktionen des im ersten Quadrupol isolierten Ions (engl. parent ion) zu ermitteln, oder selektiv nur ein bekanntes Fragmention zu beobachten. Durch das Erfassen aller Fragmentionen können Rückschlüsse auf die Struktur gezogen werden. Durch Beobachtung von nur ein oder zwei Fragmentionen, kann sehr empfindlich und selektiv quantifiziert werden. Diese Technik wird auch als Multiple Reaction Monitoring (MRM) bezeichnet.
Daneben gibt es auch andere Techniken für MS/MS (und sog. MSn). In Ionenfallen kann man ein Ion isolieren, und ihm dann entweder durch Kollision (hier aber meist mit Helium) oder auch durch Strahlung Energie zuführen. Gerade in FT-ICR-Geräten verwendet man dazu Infrarotlaser oder „Elektronenkanonen“ (engl. Electron Capture Dissociation). Eine weitere Methode ist die Electron Transfer Dissociation (ETD), die gerade im Bereich der Proteinidentifikation erste Einsatzmöglichkeiten erfährt.
Kopplung mit Chromatographieverfahren Bearbeiten
Bei sehr komplexen Proben ist es nützlich, diese mit einem vorgelegten Trennverfahren aufzutrennen, bevor man sie dem Massenspektrometer zuführt. In diesem Sinn wird Massenspektrometrie oft zusammen mit Gaschromatographie (GC/MS) oder Flüssigchromatographie (LC/MS) betrieben. Weniger weit verbreitet sind die Kopplung mit Kapillarelektrophorese (CE/MS) und Ionenmobilitäts-Spektrometrie (IMS/MS). Teilweise werden auch mehrdimensionale Trenntechniken eingesetzt z. B. GCxGC-MS. Flugzeitmassenspektrometer eignet sich besonders gut im Verbund mit mehrdimensionaler Gaschromatographie, weil damit sehr schnell Massenspektren über einen großen m/q-Bereich aufgenommen werden können. Dieses Verfahren erlaubt eine genaue Auftrennung und Detektion verschiedener Verbindungsklassen aus komplexen Matrices (z. B. Erdölproben). Dafür werden zwei GC-Säulen mit unterschiedlicher Polarität hintereinander geschaltet.
Auswertung der Massenspektren Bearbeiten
Voraussetzung für die Bestimmung der Masse m ist die Kenntnis der Ladung q des Ions, denn die Analysatoren können die Ionen nur nach dem Verhältnis m/q trennen. q ist jedoch immer ein ganzzahliges Vielfaches der Elementarladung e: q = z·e, und meistens ist z = +1 (einfach positiv geladen). Als Einheit von m/q wurde das Thomson Th vorgeschlagen: [m/q] = Th.
Zu beachten ist, dass es sich bei Massenspektren um Histogramme handelt, nicht um stetige Kurven.
Zunächst muss die Masse des Analyten bestimmt werden. Normalerweise ist das die Masse des schwersten detektierten Ions (Molekülpeak oder Molekülion). Allerdings ist bei der Elektronen-Ionisation oft ein Großteil der Moleküle gespalten. Testweise kann die Elektronenenergie verringert werden, so dass weniger Moleküle gespalten werden und der Molekülpeak deutlicher sichtbar wird.
Die weitere Auswertung basiert darauf, dass die Atome der verschiedenen chemischen Elemente einen unterschiedlichen Massendefekt haben. Daher kann aus einer sehr exakt bestimmten Masse eine Liste möglicher Summenformeln angegeben werden. Bei leichten Molekülen gibt es nur eine oder wenige passende Elementarzusammensetzungen. Mit steigender Masse oder zunehmender Anzahl an Heteroatomen steigt auch die Anzahl möglicher Kombinationen stark an.
Bei schwereren Molekülen stehen deshalb sehr viele mögliche Summenformeln zur Auswahl. Weitere Hinweise liefern die Isotopenzusammensetzungen der verschiedenen Elemente. So besteht der Kohlenstoff zum Beispiel zu 98,9 % aus 12C und zu 1,1 % aus 13C. Je nachdem, wie viele C-Atome im Molekül vorhanden sind, sind neben dem Hauptsignal im Spektrum Nebensignale zu finden, die vom Hauptpeak um 1 Th, 2 Th etc. entfernt sind und ein charakteristisches Intensitätsverhältnis zum Hauptsignal haben. Halogene wie Chlor und Brom haben ebenfalls charakteristische Isotopenverhältnisse, die zur Identifizierung benutzt werden können.
Die genannten Methoden sind auch auf die Bruchstücke anwendbar. Moleküle brechen oft an charakteristischen Stellen. Aus der Masse der Bruchstücke und evtl. weiteren Informationen kann schließlich die Strukturformel bestimmt werden.

Dabei helfen vor allem bei mit positiver EI-Ionisierung erstellten Massenspektren auch Massenspektrenbibliotheken. Die bekanntesten sind unter den Kürzeln ihrer Vertreiber die Wiley- und die NIST-Massenspektrenbibliotheken. Durch den Peak Counting Score kann eine Identifizierung erfolgen.
Die Quantifizierung von Verbindungen wird bei der Massenspektrometrie dadurch erleichtert, dass bei der Analytik isotopenmarkierte (13C-markierte oder deuterierte) interne Standards verwendet werden können.
Ein Problem bei der Datenauswertung stellen die proprietären Datenformate der einzelnen Gerätehersteller dar [18]. Die Daten werden in eigenen Binärdatenformaten vorgehalten. Meist werden vom jeweiligen Hersteller in die eigene Steuerungs- und Managementsoftware integrierte Auswertungsprogramme mitgeliefert. Um Programme von Dritten zu benutzen, bedarf es oft der Datenkonvertierung zum Datenexport, für die es im Bereich der Forschung frei erhältliche Lösungen gibt. [19]
Anwendungen Bearbeiten
Die Massenspektroskopie dient in der Analytik bzw. der analytischen Chemie als Analyseverfahren zur Bestimmung chemischer Elemente oder Verbindungen. In dieser Form werden Massenspektrometer in vielen Bereichen der Naturwissenschaften und der Technik für die Analyse von Materialien eingesetzt, unter anderem in der Chemie, Biologie, Archäologie und Klimatologie.
Auch in der Teilchenphysik werden Massenspektrometer verwendet. In diesem Bereich ist das Ziel jedoch weniger die Analyse von chemischen Elementen, sondern die Ermittlung der Massen von Elementarteilchen oder Atomkernen sowie der Detektion von noch unbekannten Teilchen.
Chemie Bearbeiten
Für einen Analyten (die zu testende Substanz) wird die Häufigkeit, mit der geladene Moleküle (Ionen) und deren Massenfragmente auftreten, bestimmt. Die Massenspektrometrie ist eine wichtige Methode der analytischen Chemie bei der Aufklärung der Struktur und Zusammensetzung von Verbindungen und Gemischen. Der qualitative (Erkennung von unbekannten Substanzen) und quantitative (wie viel Substanz einer Verbindung ist vorhanden) Nachweis sehr kleiner Substanzmengen (ca. > 10−15 g = 1 fg (Femtogramm)) ist möglich.
Archäologie Bearbeiten
Isotopenverhältnisse einiger Elemente erlauben Rückschlüsse auf die Ernährung der Menschen, deren Knochen untersucht werden. Siehe auch Isotopenuntersuchung. Das Isotopenverhältnis von Kohlenstoff 14C zu 12C im organischen Material von archäologischen Funden erlaubt es, die Zeit seit der pflanzlichen Bildung der vermessenen Substanz zu ermitteln. Zur Messung von 14C wird die Beschleuniger-Massenspektrometrie herangezogen.
Biologie Bearbeiten
Massenspektrometrie wird in der Proteomik und Metabolomik verwendet, wo die Verwendung weitgehend jener in der Chemie entspricht. Biologische Proben, insbesondere Proteine, verlangen jedoch aufgrund der Molekülgröße und der speziellen Fragestellung (Identität, Sequenz, chemische Modifikation) bei der Aufklärung von systemischen Zusammenhängen eine besondere Probenvorbereitung und Messmethodik.
Klimatologie Bearbeiten
Das Verhältnis der Häufigkeit bestimmter Isotope in Proben von Sedimenten und Eisbohrkernen lässt Rückschlüsse auf das Klima der Vergangenheit zu. Zum Beispiel verdampft Wasser, das das Isotop 16O enthält, leichter als solches, das das Isotop 18O enthält. Eiszeiten, bei denen große Mengen des Wassers als Eisschild dem Wasserkreislauf entzogen werden, verschieben die Häufigkeit dieser Isotope im Meer und damit auch im neu fallenden Schnee. Aus der Sauerstoff-Isotopenstufe kann auf die Menge des Inlandeises zu der Zeit geschlossen werden, als die Probe gebildet wurde.
Technik Bearbeiten
Die Massenspektroskopie wird auch in vielen technischen Bereichen genutzt. Sie kann beispielsweise bei der Endpunkterkennung von Ätzprozessen eingesetzt werden. Ein anderer Anwendungsbereich ist die Einstellung und Optimierung der Gaszufuhr bei Beschichtungsprozessen (genauer chemische Gasphasenabscheidung). Hierbei wird das Abgas nach der Reaktion hinsichtlich unverbrauchter Reaktionsgase untersucht und die Gaszufuhr entsprechend angepasst. Die Massenspektroskopie kann aber auch zur Analyse der abgeschiedenen Materialien eingesetzt werden. Dabei können mithilfe von SIMS auch Tiefenprofile erstellt werden, was unter anderem bei der Analyse von dünnen Schichten eingesetzt wird.
Literatur Bearbeiten
Allgemein Bearbeiten
- Herbert Budzikiewicz, Mathias Schäfer: Massenspektrometrie – Eine Einführung. Wiley-VCH, Weinheim 2005, ISBN 978-3-527-30822-4.
- Hans-Joachim Hübschmann: Handbook of GC/MS, Fundamentals and Applications. 2. rev. Ed. Wiley-VCH Verlagsgesellschaft mbH, Weinheim 2008, ISBN 978-3-527-31427-0.
- Fred W. McLafferty und Frantisek Turecek (Deutsche Übersetzung): Interpretation von Massenspektren, 4. Ed., Spektrum Akademischer Verlag GmbH, Heidelberg 1995, ISBN 0-935702-25-3.
- A. M. Boehm et al.: Command Line Tool for Calculating Theoretical MS Spectra for Given Sequences. Bioinformatics, 2004. 20(16): 2889-2891; doi:10.1093/bioinformatics/bth328.
- Alexander M. Lawson (Editor): Mass Spectrometry - Clinical Biochemistry - Principles/Methods/Applications. Walter de Gruyter & Co., Berlin/New York 1989, ISBN 3-11-007751-5.
Spektrensammlungen Bearbeiten
Spektrensammlungen liegen sowohl in gedruckten Werken als auch in gerätekompatiblen Dateiformaten vor. Letztere werden heute meist bereits mit den Geräten angeboten und für die komfortable Auswertung von unbekannten Massenspektren eingesetzt.
- M. Spiteller, G. Spiteller: Massenspektrensammlung von Lösungsmitteln, Verunreinigungen, Säulenbelegmaterialien und einfachen aliphatischen Verbindungen, Springer Verlag Wien, New York (1973), ISBN 3-211-81117-6
- A. Cornu, R. Massot: Compilation of Mass Spectral Data, Index de Spectres de Masse, Vol. 1 & Vol. 2, 2. Ed., Heyden & Son Ltd. London, Philadelphia, Rheine, (1979) ISBN 0-85501-086-X
- K. Pfleger, H. Maurer, A. Weber: Mass Spectral and GC Data of Drugs, Poisons and Their Metabolites, Part I & II, VCH Verlagsgesellschaft mbH, Weinheim (1985), ISBN 3-527-26303-9
- NIST Standard Reference Database 1A, NIST/EPA/NIH Mass Spectral Library with Search Program: (Data Version: NIST 08, Software Version 2.0f) NIST-Spektrensammlung, auch unter Einschluss von Designerdrogen verfügbar.
Quellen Bearbeiten
- W. Barger: Schulungsunterlagen zum ICP-MS Kurs 2006. LAS PerkinElmer (Germany) GmbH, Rodgau, unveröffentlicht.
- R. S. Houk, V. A. Fassel, G. D. Flesch, A. L. Gray, E. Taylor: Inductively Coupled Argon Plasma for Mass Spectrometric Determination of Trace Elements. In: Analytical Chemistry. 1980, 52, S. 2283.
- D. Skoog, J. Leary: Instrumentelle Analytik. Grundlagen, Geräte, Anwendung. Berlin 1996. (dt. Übersetzung der 4. Auflage von Principles of Instrumental Analysis, Orlando 1992).
- S. M. Nelms: ICP Mass Spectrometry Handbook. Oxford 2005.
- H. E. Taylor: Inductively Coupled Plasma-Mass Spectrometry. San Diego 2001.
- R. Thomas: Practica Guide to ICP-MS. New York, 2004.
Siehe auch Bearbeiten
Weblinks Bearbeiten
- Geschichte der Massenspektrometrie
- Massenspektrometrie – eine umfassende Darstellung der Theorie und der Technik
- Massenspektrometrie (en)
- Probenpräparation für Proteinidentifizierung mittels Nano-LC-MS
- MS-Anwendungsbeispiele in der Proteinanalytik
Kernspinresonanzspektroskopie Bearbeiten

Die Kernspinresonanzspektroskopie (NMR-Spektroskopie von englisch nuclear magnetic resonance) ist eine spektroskopische Methode zur Untersuchung der elektronischen Umgebung einzelner Atome und der Wechselwirkungen mit den Nachbaratomen. Dies ermöglicht die Aufklärung der Struktur und der Dynamik von Molekülen sowie Konzentrationsbestimmungen.
Da die Methode auf dem magnetischen Moment der Atomkerne beruht, sind nur Nuklide mit ungerader Massen- oder Ordnungszahl einer NMR-spektroskopischen Untersuchung zugänglich, da nur diese einen magnetischen Atomkern haben können. In der Organischen Chemie und Biochemie spielen 1H- und 13C-NMR-Spektroskopie eine wichtige Rolle.
Geschichte Bearbeiten

Als Ursprung der Kernspinresonanzspektroskopie kann man das Atomstrahlexperiment von Otto Stern im Jahr 1933 ansehen.[20] Er konnte zeigen, dass ein Strahl von Silberatomen durch ein Magnetfeld in zwei Teilstrahlen aufgespalten wird, die den beiden Spinzuständen zugeschrieben wurden. Stern erhielt für diese Arbeiten 1943 den Nobelpreis für Physik.
Die ersten Kernspinresonanz- und ESR(Elektronenspinresonanz)-Experimente führte Isidor Isaac Rabi, der 1944 mit dem Nobelpreis für Physik ausgezeichnet wurde, mit einer modifizierten Stern-Gerlach-Anordnung durch. Er zeigte, dass einer der Halbstrahlen verschwand, wenn man auf ihn mit Hilfe einer Spule ein elektromagnetisches Wechselfeld geeigneter Frequenz (nämlich der Larmorfrequenz) einstrahlte.[21] Das erste erfolgreiche Magnetresonanzexperiment in kondensierter Materie führte E. K. Zavoisky in Kasan an der Wolga im Jahr 1944 durch.[22] Es handelte sich allerdings um ein ESR-Experiment. 1946 veröffentlichten Felix Bloch und Edward Mills Purcell unabhängig voneinander erstmals erfolgreiche NMR-Experimente in flüssiger und fester Phase. Sie erhielten dafür den Nobelpreis für Physik im Jahr 1952.[23] Die ersten erfolgreichen NMR-Experimente in Europa führte Harry Pfeifer in Leipzig im Jahr 1951 durch.
Nachdem kurz darauf die Aufspaltung der Spektren durch chemische Verschiebung und skalare Kopplung erkannt wurde, begann die Kernspinresonanzspektroskopie sich zu einer wichtigen Methode in der chemischen Strukturaufklärung zu entwickeln. Zunächst wurde hauptsächlich die Continuous-Wave-(CW)-Methode benutzt, bei der durch Variation der Frequenz oder des Feldes die Resonanzen nacheinander angeregt wurden.[24] 1947 reichten Russell Varian und Felix Bloch ein Patent ein für das erste Kernspinresonanz-Spektrometer. Das erste kommerzielle Kernspinresonanz-Spektrometer baute 1952 die Firma Varian Associates in Palo Alto. Um 1955 baute die japanische Firma Jeol ebenfalls NMR-Spektrometer. Die US-amerikanische Biophysikerin Mildred Cohn setzte in den frühen sechziger Jahren die Kernspinresonanz-Spektroskopie zur Aufklärung metabolischer Prozesse auf molekularer Ebene ein.[25] Ihre bahnbrechenden Pionierarbeiten auf diesem Gebiet mündeten in Methoden und Anwendungen, die noch heute von vielen Forschern genutzt werden.

Da die CW-Technik durch ein schlechtes Signal-Rausch-Verhältnis gekennzeichnet war, entwickelte ab Mitte der 1960er Jahre Richard R. Ernst (Nobelpreis für Chemie 1991) bei der Firma Varian ein Puls-Fourier-Transformation-NMR-Spektrometer (FT-NMR), das eine wesentlich schnellere Aufnahme der Spektren ermöglichte, was – bei gleicher Messzeit – im Vergleich zu den CW-Spektrometern eine wesentliche Steigerung der Empfindlichkeit und damit des Signal-Rausch-Verhältnisses bedeutete.[26] Bereits in den Jahren 1949 und 1950 wurden von Hahn und Torrey die ersten Pulsverfahren untersucht.[27] Die ersten kommerziellen Kernspinresonanz-Impulsspektrometer wurden Mitte der 1960er Jahre von der deutschen Firma Bruker[28] (gegründet von Günther Laukien, einem der NMR-Pioniere in Deutschland) in Karlsruhe von einer Gruppe um Bertold Knüttel und Manfred Holz gebaut. Es folgte die Einführung von Breitbandentkopplung und von Mehrpulsverfahren. Nach einer Idee von Jean Jeener wurden ab Anfang der 1970er Jahre Mehrpulsexperimente mit einer systematisch variierten Wartezeit zwischen zwei Pulsen entwickelt, die nach Fourier-Transformation über zwei Zeitbereiche zu zweidimensionalen Spektren führten.
Kurt Wüthrich und andere bauten diese 2D- und Multi-Dimensions-NMR zu einer bedeutenden Analysetechnik der Biochemie aus, insbesondere zur Strukturanalyse von Biopolymeren wie Proteinen. Wüthrich bekam für diese Arbeiten 2002 den Nobelpreis in Chemie.[29] Im Gegensatz zur Röntgenstrukturanalyse liefert die Kernspinresonanz-Spektroskopie Strukturen von Molekülen in Lösung. Von besonderer Bedeutung ist die Möglichkeit, detaillierte Informationen über die Moleküldynamik mit Hilfe von Relaxationsparametern zu gewinnen.
Wissenschaftlicher Hintergrund Bearbeiten
Quantenmechanische Grundlagen Bearbeiten

Teilchen und Atomkerne, die einen Kernspin besitzen, haben als rotierende Ladungsträger ein magnetisches Moment, das oft mit bezeichnet wird. Das magnetische Moment von Atomkernen kann in einem äußeren Magnetfeld nicht jede beliebige, sondern nur bestimmte, durch die Quantenmechanik beschriebene Orientierungen einnehmen. Die Zahl der möglichen Orientierungen wird durch die Kernspinquantenzahl bestimmt (siehe: Multiplizität). Zu jeder Kernspinquantenzahl existieren Orientierungen und jeder Orientierung ist eine magnetische Kernspinquantenzahl zugeordnet.





Beispiele:
- Wasserstoff-Kern mit Kernspin ½ : zwei Orientierungen mit +½ und −½
- Deuterium-Kern mit Kernspin : drei Orientierungen mit +1, 0 und -1



Ohne ein äußeres Magnetfeld sind die mit gekennzeichneten Zustände energetisch gleich (siehe Entartung (Quantenmechanik)). In Anwesenheit eines äußeren Magnetfeldes entstehen Energiedifferenzen (Zeeman-Effekt). Kernresonanz-Phänomene beruhen auf der Anregung von Kernspin-Übergängen zwischen solchen -Zuständen. Die dazu benötigte Energie ist proportional zur Stärke des äußeren Magnetfeldes und zum gyromagnetischen Verhältnis des betrachteten Atomkerns:



Diese Energie wird durch Einstrahlen von resonanten elektromagnetischen Wellen eingebracht. Die Resonanzfrequenz wird in der NMR als Larmor-Frequenz bezeichnet und liegt im Radiowellen-Bereich. Gängige NMR-Spektrometer arbeiten bei Protonen-Resonanzfrequenzen zwischen 300 MHz und 1 GHz.

Wenn alle 1H- oder 13C-Atome die exakt gleiche Larmor-Frequenz hätten, wäre die NMR-Methode zur Strukturaufklärung wenig interessant. Tatsächlich hängen aber die Resonanzfrequenzen von den individuellen, atomar aktiven Magnetfeldern ab. Diese lokalen Magnetfelder können in ihrer Stärke vom Hauptmagnetfeld abweichen, beispielsweise durch den Einfluss der elektronischen Umgebung eines Atomkerns oder durch magnetische Wechselwirkung zwischen benachbarten Atomkernen. Aufgrund dieser Eigenschaften wird die Kernresonanzspektroskopie zur Strukturaufklärung von Molekülen eingesetzt.
Messverfahren der Kernresonanzspektroskopie Bearbeiten
Zur Messung bringt man die Probe in ein homogenes, magnetisches Feld. Heutzutage werden solche sog. Hauptmagnetfelder mit Hilfe von supraleitenden Elektromagneten erzeugt, die mit flüssigem Helium und Stickstoff gekühlt werden. Die Probe wird von einer Induktionsspule umgeben, welche ein hochfrequentes elektromagnetisches Wechselfeld senkrecht zum Hauptmagnetfeld erzeugt. Dann variiert man die Stärke des Hauptmagnetfeldes, bis der Resonanzfall eintritt (Continuous-Wave-Verfahren, veraltet). Alternativ kann auch die magnetische Feldstärke konstant gehalten und die Frequenz des eingestrahlten Wechselfeldes variiert werden (engl. continuous field, veraltet). Wenn der Resonanzfall eintritt, die Probe also Energie aus dem Wechselfeld aufnimmt, verändert sich die Stromstärke, welche zum Aufbau des Wechselfeldes benötigt wird. Dies kann man messen.
Moderne Messverfahren strahlen nicht mehr kontinuierliche Wechselfelder in die Probe ein, sondern Radiowellen-Pulse. Ein kurzer Radiowellenpuls regt dabei ein Frequenzband an, dessen Frequenzbreite über die Fourier-Beziehung umgekehrt proportional zur Pulsdauer ist. Dadurch werden in der Probe alle Übergänge, die in dieses Frequenzband fallen, gleichzeitig angeregt. Bei korrekter Wahl von Pulsdauer und Pulsleistung kann die Magnetisierung der angeregten Kernspins in die Transversalebene senkrecht zum Hauptmagnetfeld gebracht werden. Nach Beendigung des Pulses oszilliert diese Transversalmagnetisierung für kurze Zeit senkrecht zum Hauptmagnetfeld. Dabei oszilliert jeder Kernspin mit seiner individuellen Larmor-Frequenz. Diese Summe dieser Oszillationen wird als elektrischer Strom über elektromagnetische Induktion mit der gleichen Induktionsspule detektiert, die auch zum Senden des Anregungspulses gedient hatte. Das empfangene Signal wird digitalisiert und aufgezeichnet. Mit Hilfe der schnellen Fourier-Transformation ist es möglich, die individuellen Larmor-Frequenzen aus der Summe der Oszillationen zu extrahieren, um ein NMR-Spektrum zu erhalten. Darum tragen moderne NMR-Verfahren den Namen PFT-NMR für Pulsed Fourier Transform NMR Spectroscopy.
Relaxation Bearbeiten
Bei der PFT-NMR existieren drei Relaxationsprozesse, die einerseits die Leistungsfähigkeit der PFT-NMR einschränken, aber andererseits einzigartige Informationen zur Molekül-Dynamik und Material-Inhomogenitäten liefern können. Die -Relaxation ist der Prozess der Rückkehr der Kernspins vom angeregten Zustand zum thermischen Gleichgewicht unter Abgabe der bei der Anregung aufgenommenen Energie als Wärme. Wird zwischen zwei NMR-Experimenten (scans) nicht die vollständige -Relaxation abgewartet, so steht für jedes weitere nur eine gewisse, geringere Magnetisierung und damit Signalintensität zur Verfügung. Falls eine kurze Repetitionzeit gewünscht ist, muß auch der Anregungswinkel (auf den sog. Ernst-Winkel) verkleinert werden, um trotzdem ein möglichst starkes Signal zu erhalten. Die -Relaxation ist die Dephasierung der Transversalmagnetisierung aufgrund entropischer Effekte, die mit der magnetischen Dipol-Dipol-Wechselwirkung benachbarter Atomkerne zusammenhängt. Hier wird keine Energie abgegeben, da die Spins im angeregten Zustand verbleiben, aber die Transversalmagnetisierung läuft von einem Vektor zunächst zu einem Fächer und zuletzt zu einer Kreisfläche auseinander, so dass kein NMR-Signal mehr in der Detektionsspule induziert wird. Befinden sich zusätzlich Magnetfeldinhomogenitäten in der Probe, sei es durch Imperfektionen des Hauptmagnetfeldes oder durch Suszeptibilitätsunterschiede innerhalb der Probe, wird statt der -Relaxation die beschleunigte -Relaxation beobachtet.



Die -Relaxation bzw. -Relaxation beschränkt die Lebensdauer des NMR-Signals direkt nach der Anregung. Das NMR-Signal wird darum als gedämpfte Schwingung, als FID (free induction decay) gemessen. In der Praxis wird die -Relaxationszeit mit Hilfe der Spin-Echo-Methode gemessen.
Die -Relaxation limitiert, wie schnell man NMR-Experimente hintereinander ausführen kann. Die -Relaxation und die -Relaxation hängen stark von der Dichte und Viskosität/Rigidität der Probe ab.
Empfindlichkeit der NMR-Spektroskopie Bearbeiten
Die Energiedifferenzen der -Zustände sind sehr klein, und obwohl die energetisch günstigeren Zustände bevorzugt besetzt werden, sind die Populationsunterschiede zwischen den Zuständen im thermischen Gleichgewicht sehr gering (Boltzmann-Statistik). Darum können nur wenige Kernspin-Übergänge angeregt werden und die detektierten NMR-Signale sind relativ schwach. Dieses Problem kann auf verschiedene Arten umgangen werden:
- Senkung der Temperatur
- Erhöhung der Magnetfeldstärke
- Anwendung von Hyperpolarisation
- Optimierung der Signal-Detektion
- Signal-Akkumulation
Die Senkung der Temperatur und die Erhöhung der Magnetfeldstärke ändern das thermische Besetzungsgleichgewicht der -Zustände, so dass mehr Kernspin-Übergänge angeregt werden können. Mit Hilfe der Hyperpolarisation kann ein Besetzungs-Ungleichgewicht erzeugt werden, das stark vom thermischen Gleichgewicht abweicht und in dem der energetisch günstigste -Zustand fast vollständig besetzt ist.
Zur Optimierung der Signal-Detektion wird rauscharme Elektronik verwendet. Der Einsatz von elektrischen Schwingkreisen begrenzt die Detektion auf ein schmales Frequenzband im Bereich der erwarteten Larmor-Frequenz, d.h. die Detektion von Störsignalen und von Rauschen aus anderen Frequenzbereichen wird unterdrückt.
Die Signal-Akkumulation dient dazu, das Signal-Rausch-Verhältnis zu verbessern. Eine NMR-Messung wird -mal auf identische Weise durchgeführt und die gemessenen Signale der einzelnen Messungen werden addiert. Durch diese Akkumulation nimmt die NMR-Signalstärke um den Faktor zu, während statistisches Rauschen nur um den Faktor zunimmt. Da zwischen NMR-Experimenten die vollständige -Relaxation der Spins erfolgen sollte und da die -Relaxation organischer Substanzen einige zehn Sekunden dauern kann, kann die Signal-Akkumulation zu einer erheblichen Verlängerung der Messdauer führen.


Auflösungsvermögen Bearbeiten
Die erreichbare Auflösung eines Pulsspektrometers ist invers proportional zu Länge des FID-Signals. Neben der transversalen Relaxation ( ) der Probe wird sie von der Inhomogenität des -Feldes in der Probe bestimmt. Das Ausgleichen von Magnetfeld-Inhomogenitäten erfolgt über das sog. Shimming. Dazu werden im Spektrometer mit Hilfe elektrischer Ströme schwache Magnetfelder zusätzlich zum Hauptmagnetfeld erzeugt, mit denen lokale Inhomogenitäten zum Teil ausgeglichen werden können. Die erhöhte Homogenität des resultierenden Gesamtmagnetfeldes reduziert die -Relaxation, wodurch das NMR-Signal langlebiger wird.
Anwendungsgebiete Bearbeiten
NMR-Spektren können am einfachsten für Moleküle aufgenommen werden, die sich in Lösung befinden und nicht mit paramagnetischen Substanzen in Wechselwirkung stehen. NMR-Spektroskopie an paramagnetischen Substanzen und an Festkörpern ist ebenfalls möglich, die Interpretation der Spektren und die Aufbereitung der Proben für die Messung sind aber in beiden Fällen deutlich komplexer. Bezüglich der NMR an Festkörpern vgl. auch Magic-Angle-Spinning.
Die hochauflösende Kernresonanzspektroskopie in Lösung wird heute in großem Maßstab für folgende Aufgaben verwendet:
- Zum zerstörungsfreien Nachweis von Inhaltsstoffen einer Probe
- Zur Bestimmung von Molekülstrukturen (von kleinen Molekülen bis hin zu Proteinen und Nukleinsäurefragmenten)
- Zur Untersuchung von Wechselwirkungen zwischen Molekülen
Neben spektroskopischen Untersuchungen vermittelt auch die Bestimmung von Kernspin-Relaxationszeiten Informationen über die Struktur und Dynamik von Materialien. In Flüssigkeiten z.B., deren Mikrostruktur und -Dynamik mit herkömmlichen Methoden nur schwer erforscht werden kann, können Abstände zwischen molekularen Nachbarn und molekulare Umorientierungszeiten, die typischerweise im Pico- bis Nanosekunden- Bereich liegen, mittels Relaxationszeitmessungen bestimmt werden.
Unterschiedliche Kernspin-Relaxationszeiten in verschiedenen biologischen Geweben bilden auch die Basis für die in der Medizin als bildgebendes diagnostisches Verfahren genutzte Magnetresonanztomographie (Kernspintomographie). Magnetresonanztomographie-Methoden finden außer in der medizinischen Diagnostik auch zunehmend Anwendungen in den Ingenieur- und Geowissenschaften.
Ein weiteres wichtiges Anwendungsgebiet ist die Untersuchung der translatorischen Moleküldynamik, also von Diffusion und Fließbewegungen von Molekülen oder Molekülaggregaten in Flüssigkeiten und Festkörpern mittels Feldgradienten-NMR.[30] Mit der sogenannten diffusion-ordered-spectroscopy (DOSY) kann in Mischungen die translatorische Beweglichkeit der, NMR-spektroskopisch identifizierten, Einzelkomponenten gemessen werden.
Methoden zur Strukturaufklärung in der organischen Chemie Bearbeiten
Chemische Verschiebung und Integration von Signalen Bearbeiten
Die Resonanzfrequenzen werden nicht als Absolutwerte, sondern als chemische Verschiebung gegenüber einer Referenzsubstanz, dem so genannten Standard, angegeben. Die chemische Verschiebung ist definiert als
- ,

wodurch sie unabhängig von der Feldstärke des gerade verwendeten Magneten wird. Da die Werte der chemischen Verschiebung sehr klein sind, werden sie in ppm angegeben. Als Standard für 1H- und 13C-Spektren organischer Lösungen wird die Resonanzfrequenz der jeweiligen Kerne in Tetramethylsilan (TMS) verwendet. Während früher jeder Probe einige Milligramm TMS zugesetzt wurden, bezieht man sich heutzutage in der Regel auf die bekannte chemische Verschiebung der Restprotonen des deuterierten Lösungsmittels.
Die chemische Verschiebungen von Wasserstoffkernen in organischen Molekülen wird durch die Art der funktionellen Gruppen beeinflusst. Je nach der Struktur des Moleküls weichen die chemischen Verschiebungen gleicher funktioneller Gruppen leicht voneinander ab, so dass das NMR-Spektrum charakteristisch für eine Substanz ist. Außerdem werden sie auch durch benachbarte Moleküle in der Probe beeinflusst, so dass in unterschiedlichen Lösungsmitteln oder in der Reinsubstanz unterschiedliche relative und absolute Resonanzfrequenzen der Protonen einer Probe auftreten. Bei starken Wechselwirkungen zwischen Substanz und Lösungsmittel treten dabei auch Unterschiede von mehreren ppm auf. Mit Hilfe der Shoolery-Regel kann die chemische Verschiebung abgeschätzt werden.
Die relative Anzahl der einem bestimmten Signal zugrundeliegenden Wasserstoffatome ist bei einfacher 1H-Spektroskopie proportional zum Flächeninhalt (dem Integral) des betreffenden Signals.
Durch Auswertung dieses Integrals kann also beispielsweise bestimmt werden, wie viele Wasserstoffatome eines Moleküls sich an Methylgruppen, an Aromaten, an Carboxygruppen, an Doppelbindungen usw. befinden. Diese Kenntnis ist für die organische Chemie bei der Bestimmung von Strukturen äußerst wichtig.
| Wasserstoffatome | typische chemische Verschiebungen [ppm] |
|---|---|
| H3C– | 0,9 |
| H3CCR=C | 1,6 |
| H3C–Ar | 2,3 |
| H3C–CO–R | 2,2 |
| H3C–OR | 3,3 |
| R2C–CH2–CR2 | 1,4 |
| –C–CH2–Cl | 3,6 |
| Ar-H | 6,8 bis 7,5 (bis 8.5 bei Heteroaromaten) |
| R-CHO | 9 bis 10 |
| R–COOH | 9 bis 13 |
| ROH | 0,5 bis 4,5 |

Skalare Kopplung Bearbeiten
Durch die skalare Kopplung werden die Spinzustände durch in ihrer Nachbarschaft befindliche weitere Kerne mit von Null verschiedenem Spin unabhängig vom anliegenden externen Feld in energetisch unterschiedliche Niveaus aufgespalten, deren Zahl von der Anzahl möglicher unterschiedlicher Orientierungen der einzelnen Spins abhängt. Diese Kopplung wird durch die Spins der die Bindung zwischen den Teilchen bildenden Elektronenpaare vermittelt, ihre Wirkung ist gewöhnlich über bis zu vier Bindungen feststellbar. Kerne, die chemisch und spin-symmetrisch gleich sind, koppeln nicht miteinander (sie sind magnetisch äquivalent).
Aus dem dadurch beobachteten Kopplungsmuster kann der Spektroskopiker die Nachbarschaftsverhältnisse der einzelnen Kerne, und damit in vielen Fällen die vollständige Struktur einer Verbindung erschließen.
Mitunter kann es hilfreich sein, für die Strukturaufklärung eine bestimmte oder alle Kopplungen zu unterdrücken. Dazu wird ein Radiosignal mit der Frequenz eines Kernes oder einer ganzen Gruppe von Kernen (Breitbandentkopplung) eingestrahlt, das übrige Spektrum verhält sich dann so als wäre der entsprechende Kern nicht vorhanden, da dessen Besetzungsunterschied dann null ist. 13C-Spektren werden standardmäßig 1H-entkoppelt, da sie durch die Überlappung der Kopplungsmuster der einzelnen Kerne sonst oft uninterpretierbar wären. Außerdem wird die geringe Empfindlichkeit der Methode durch die fehlende Aufspaltung verbessert.
In anisotropen Proben (Substanzen mit kristallinen Anteilen) wird der richtungsabhängige Teil der skalaren Kopplung, sowie die dipolare Kopplung, nicht mehr zu null ausgemittelt. Solche Proben zeigen große, feldunabhängige Aufspaltungen, bzw. Linienverbeiterungen von mehreren kHz für 1H-Spektren.
Funktionsprinzip Bearbeiten
Die Kernsuszeptibilitäten sind wesentlich geringer als die diamagnetischen und paramagnetischen Suszeptibilitäten (Faktor:104). Bei Dia- und Paramagnetismus sind die Elektronen des Atoms für die Suszeptibilität verantwortlich. Bei Kernen kann die Suszeptibilität mit der Langevin-Debye-Formel bestimmt werden.
Früher wurden NMR-Resonanzen mit einer Brückenschaltung in Schwingkreisen bestimmt. Bloch und Mitarbeiter nutzten zwei identische Schwingkreise, d. h. zwei Spulen und zwei Kondensatoren, um einen Abgleich mit einer Brückenschaltung vorzunehmen; eine Spule als Sender eine als Empfänger. Es ist aber auch möglich, eine Brückenschaltung mit nur einer Spule herzustellen. Dieses Verfahren wurde von Purcell genutzt.
Vor der Probenvermessung wird die Brücke mit der zu messenden Frequenz abgeglichen. Mit Gleichungen aus der Physik kann man für einen Schwingkreis und eine Brückenschaltung die Phasenverschiebung zwischen Strom und Spannung, den Scheinwiderstand und die Stromlosigkeitsbedingungen einer Brücke berechnen.
In die Spule kommt nun ein Substanzröhrchen hinein. Ein Magnetfeld (mit einem Permanentmagneten oder Elektromagneten) wird dann horizontal zur Spulenachse erzeugt. Bei einer ganz bestimmten Frequenz und einer bestimmten Magnetfeldstärke und nur bei Anwesenheit einer Substanzprobe (mit entsprechenden Atomkernen) wird der Schwingkreis verstimmt. Im Oszilloskop oder mit einem Schreiber ist diese Verstimmung sichtbar.
Sehr bedeutsam war die Bestimmung der räumlichen Magnetisierung durch das angelegte Magnetfeld. Bloch führte für alle Raumrichtungen Berechnungen durch und konnte die schwingungsabhängigen Suszeptibilitäten für die Raumrichtungen ableiten (Bloch-Gleichungen). Ungeklärt blieb jedoch noch die Frage der Relaxationszeit, das heißt, der Zeitdauer bis der angeregte Kernspin auf das Grundniveau zurückfällt. Mittels paramagnetischer Salze konnte dann die Relaxationszeit von reinen Wasserstoffprotonen auf etwa drei Sekunden berechnet werden.
Wasserstoffkerne konnten auch bei sehr geringen Frequenzen (wenige kHz) und einem sehr schwachen Magnetfeld durch Schwingkreisverstimmung nachgewiesen werden. Interessant wird die Methode für die Strukturaufklärung von komplexen Molekülen jedoch erst bei hohen Frequenzen (ab 60 MHz) und stärkeren Magnetfeldern (1,4 Tesla), da sich dann die chemischen Verschiebungen von unterschiedlichen Wasserstoffatomen komplizierter Verbindungen deutlicher unterscheiden. Will man jedoch nicht nur ein einziges Signal auf dem Oszilloskop sehen, sondern mehrere unterschiedliche Wasserstoffatomkerne (oder andere Kerne) so muss ein ganzes Frequenzband eingestrahlt werden.
Früher – bis in die 1970er Jahre – nutzten die NMR-Spektrometer das Continuous-Wave-Verfahren (CW), um das Spektrum einer komplexen Verbindung abzutasten.
Heute ist die Puls-Fourier-Transformation (PFT) üblich. Hierbei wird ein Hochfrequenzimpuls eingestrahlt. Dieser Impuls enthält ein ganzes Band an Schwingungen.
Die bereits angesprochene Abhängigkeit der Energieniveaus der Kernspins von der Molekülstruktur rührt in erster Linie von der Wechselwirkung der Elektronenstruktur der Moleküle mit dem äußeren Magnetfeld her: Hierdurch entsteht in der Elektronenhülle ein Induktionsstrom, welcher wiederum ein Magnetfeld erzeugt, das dem äußeren entgegen gerichtet ist. Dadurch wird das Magnetfeld am Atomkern geschwächt, die Frequenz der für den Übergang notwendigen Strahlung ist also kleiner als im Falle eines nackten Atomkerns. Die Differenz heißt chemische Verschiebung und wird üblicherweise im Verhältnis zur für den nackten Atomkern nötigen Frequenz angegeben. Chemische Verschiebungen liegen üblicherweise im Bereich von 0–5000 ppm.
Das magnetische Feld wird am Atomkern durch die Ausrichtung weiterer magnetischer Momente in der unmittelbaren Umgebung beeinflusst. Befindet sich beispielsweise ein Kern mit zwei Ausrichtungsmöglichkeiten in der Nähe, so kann dieser das Feld verstärken oder abschwächen. Dies führt zu einer Aufspaltung des Signals, man spricht von einer Kopplung. Weil die chemische Verschiebung im wesentlichen von der Elektronendichte am Atomkern abhängt, kann man für Atomkerne in chemisch ähnlichen Umgebungen auch ähnliche Verschiebungen erwarten. Aus der Kopplung erhält man zusätzlich Informationen über Nachbarschaftsbeziehungen zwischen verschiedenen Kernen in einem Molekül. Beides zusammengenommen liefert wesentliche Hinweise über die Struktur des gesamten Moleküls.
Atomkerne mit einer ungeraden Protonen- und/oder Neutronen-Zahl besitzen einen Kernspin I. Dieser kann ganz- und halbzahlige Werte (z. B. 1/2, 1, 3/2, …, 9/2) annehmen: bei den sogenannten uu-Kernen ist I = n (also nur ganzzahlig: 1,2,3,...) während bei gu- und ug-Kernen I = (2n+1)/2 ist (also halbzahlig: 1/2, 3/2, 5/2,...), bei Isotopen mit gerader Protonen- und Neutronenzahl (sogenannten gg-Kernen) ist I = 0. Von Null verschiedene Kernspins gehen mit einem magnetischen Dipolmoment einher. Die Größe dieses Dipolmoments wird durch das gyromagnetische Verhältnis des betreffenden Isotops beschrieben. In einem äußeren, statischen Magnetfeld richten sich magnetische Kernmomente entsprechend den Regeln der Quantenmechanik aus. Ein Atomkern mit I = ½ hat die Form einer Kugel, Kerne mit I > ½ haben eine ellipsoidische Form und haben daher zusätzlich ein elektrisches Quadrupolmoment „eQ“, welches mit elektrischen Feldgradienten wechselwirken kann (siehe auch Kernquadrupolresonanz-Spektroskopie). Diese zusätzliche starke, elektrische Wechselwirkungsmöglichkeit führt zu breiten NMR-Resonanzlinien, die komplizierter zu interpretieren sind als die schmalen, durch gut auflösbare Kopplungen strukturierten Resonanzlinien der Spin-½-Kerne.
Die am meisten für die chemische Strukturaufklärung genutzten Isotope sind daher Kerne mit Spin ½. Hierzu gehören unter anderem die Nuklide 1H, 13C, 15N, 19F, 29Si und 31P. Spin-½-Kerne können nur zwei diskrete Zustände annehmen, nämlich entweder parallel oder antiparallel zum äußeren Magnetfeld. Zwischenstellungen sind quantenmechanisch verboten. Die zwei Anordnungsmöglichkeiten entsprechen zwei unterschiedlichen Energiezuständen.
Die Energiedifferenz zwischen diesen beiden Zuständen ist proportional zur Stärke des Magnetfelds am Kernort. Der Proportionalitätsfaktor ist dabei das gyromagnetische Verhältnis des betreffenden Isotops. Übergänge zwischen den beiden Orientierungen der Kernmomente können durch die Einstrahlung resonanter magnetischer Wechselfelder ausgelöst werden. Die Resonanzfrequenz ist der Energieaufspaltung zwischen den beiden Kernspins proportional und wird auch als Larmorfrequenz bezeichnet.

Veranschaulichen lässt sich dies durch das nebenstehende Diagramm. Hierbei denkt man sich ein Koordinatensystem mit dem äußeren Magnetfeld entlang der z-Achse. Ein Atomkern mit einem Spin von ½ richtet sich mit einem Spin-Vektor entweder parallel oder antiparallel zum äußeren Feld aus. Wenn man nun die Vektoren mehrerer Atome in dieses Koordinatensystem aufnimmt, entstehen zwei Kegel, jeweils einer für parallel und antiparallel. Infolge des Energie-Unterschieds zwischen der parallelen und der antiparallelen Orientierung der magnetischen Kernmomente gibt es im thermischen Gleichgewicht einen Besetzungsunterschied zwischen den beiden Orientierungen. Dieser folgt in Hochtemperatur-Näherung der Boltzmann-Verteilung und bewirkt eine Überschussmagnetisierung in positiver Richtung entlang der z-Achse.
Das NMR-Signal kommt dadurch zustande, dass man die zu untersuchende Probe im Magnetfeld einem Radiofrequenz-Puls aussetzt. Dabei werden die Spins der einzelnen Atome durch das Magnetfeld des Pulses beeinflusst, so dass der Gesamtvektor, der sich aus den gezeigten Spin-Kegeln ergibt, gekippt wird. Er liegt nun nicht mehr parallel zur z-Achse, sondern ist um einen Winkel ausgelenkt, der proportional zur Dauer und Intensität des Radiofrequenzpulses ist. Typisch sind Pulslängen von etwa 1–10 µs. Eine maximale Quermagnetisierung senkrecht zur z-Achse wird bei einem Auslenkungswinkel von 90° erreicht.
Diese Quermagnetisierung verhält sich wie ein magnetischer Kreisel und präzediert in der Ebene senkrecht zum statischen Magnetfeld. Diese Präzessionsbewegung macht sich als sehr schwaches magnetisches Wechselfeld bemerkbar, das mittels geeigneter Verstärker gemessen wird. Nach Beenden der resonanten Einstrahlung treten zwei Prozesse, sogenannte Relaxationsprozesse, ein, durch die die Quermagnetisierung wieder abnimmt. Das NMR-Signal wird also nach Beenden des Radiofrequenzpulses als Freier Induktionszerfall (FID; von englisch: free induction decay) gemessen. Die Zeitkonstante dieses freien Induktionszerfalls hängt von der transversalen Relaxationszeit sowie von der Homogenität des Magnetfelds ab. Für leicht bewegliche Flüssigkeiten in homogenen Magnetfeldern kann sie im Bereich von mehreren Sekunden liegen. Der FID wird durch die Frequenzunterschiede infolge von chemischer Verschiebung und Kopplung moduliert. Durch eine Fourier-Transformation kann die Verteilung der verschiedenen Frequenzen aus dem FID berechnet werden. Dies ist dann das NMR-Spektrum. Das NMR-Spektrum liefert in vielen Fällen einen eindeutigen „Fingerabdruck“ des jeweiligen Moleküls. Zusammen mit Informationen aus weiteren Messungen wie z. B. der Massenspektrometrie kann aus den Spektren die chemische Struktur einer unbekannten Substanz bestimmt werden.
Kommerzielle NMR-Spektrometer für die chemische Strukturaufklärung arbeiten üblicherweise bei magnetische Flussdichten zwischen 7 und 21 Tesla. Für 1H entsprechen die Resonanzfrequenzen (Larmorfrequenzen) dann zwischen 300 und 900 MHz. Da 1H der wichtigste NMR-Kern ist, wird die Feldstärke von Spektrometern gewöhnlich in dessen Larmorfrequenz ausgedrückt. Bei 1H beträgt die Aufspaltung der Spektren infolge unterschiedlicher chemischer Verschiebungen ca. 10 ppm. Dies entspricht also einer maximalen Bandbreite von ca. 3 kHz bei einer NMR-Frequenz von 300 MHz. Die Frequenzbandbreite der NMR-Spektren infolge der chemischen Verschiebung wächst proportional zum Magnetfeld an. Die chemische Verschiebung selbst ist als Verhältnis der Differenz der Resonanzfrequenz des Kerns in einer bestimmten chemischen Umgebung und der Resonanzfrequenz in einer Referenzverbindung zur Resonanzfrequenz selbst definiert. Dies erlaubt einen einfachen Vergleich zwischen NMR-Spektren, die bei verschiedenen Feldern gemessen wurden. Für Wasserstoff und Kohlenstoff wird Tetramethylsilan (TMS)) als Referenzsubstanz genommen. Der Bereich von chemischen Verschiebungen für Kohlenstoff und viele andere Kerne ist wesentlich breiter als für Wasserstoff und kann mehrere 100 ppm betragen. Bei einigen sehr schweren Kernen wie z. B. 207Pb werden auch chemische Verschiebungen im Bereich von Prozent beobachtet.
Empfindlichkeit Bearbeiten
Ein inhärentes Problem der NMR-Spektroskopie ist ihre vergleichsweise geringe Empfindlichkeit (schlechtes Signal-Rausch-Verhältnis). Für Messungen sind je nach Experiment und Messzeit ca. 10 nmol bis 1 µmol Substanz notwendig (typische Probenmenge: 1 mL einer Lösung mit einer Konzentration von 10 µmol/L bis 1 mmol/L).
Ursache dafür sind die durch die Boltzmannverteilung festgelegten geringen Besetzungsunterschiede der Energieniveaus:

Mit dieser Gleichung wird das Besetzungsverhältnis der beiden beteiligten Energiezustände durch deren Energiedifferenz im Verhältnis zur thermischen Energie bei gegebener Temperatur T ausgedrückt. Darin ist k die Boltzmann-Konstante. Die Energiedifferenz entspricht dabei der Energie eines Lichtquants ( ), das ein Teilchen vom günstigeren in den ungünstigeren Zustand befördert (Grundgleichung der Spektroskopie). Bei einer Resonanzfrequenz von 600 MHz und einer Temperatur von 0 °C bzw. 273 K ergibt sich ein Wert von ungefähr e0,0001, also in etwa gleich eins. Daher sind schon im thermischen Gleichgewicht fast gleich viele Kerne im angeregten Zustand wie im Grundzustand. Zum Vergleich: Sichtbares Licht besitzt um einen Faktor von etwa 1 Million höhere Frequenzen. Folglich haben Übergänge, die durch sichtbares Licht angeregt werden, Besetzungsunterschiede von etwa e100, liegen also vollständig im Grundzustand vor, was die Spektroskopie im sichtbaren Bereich wesentlich empfindlicher macht.


Um die Empfindlichkeit zu steigern, werden verschiedene Wege eingeschlagen:
- Messung möglichst empfindlicher Kernsorten (besonders 1H)
- Mehrfache Messung einer Probe und Addition aller Spektren
- Einsatz stärkerer Magneten (supraleitende Magnete).
- Elektronisches Rauschen durch Kühlung der Empfänger verringern (Cryoelektronik).
- Anreicherung magnetischer Kerne, deren natürliche Häufigkeit gering ist (z. B. 13C bzw. 15N). Das wird z. B. bei Proteinen oft gemacht.
- Verwendung von Hyperpolarisationsmethoden, um die Besetzungsunterschiede künstlich zu vergrößern.
- Ausnutzen des Kern-Overhauser-Effekts (NOE)
- Übertragung der Magnetisierung empfindlicher Kerne (1H) auf unempfindlichere (13C (Kreuzpolarisation CP))
Puls-Fourier-Transform NMR Bearbeiten

Heutzutage arbeiten alle modernen NMR-Spektrometer mit der Puls-Technik. Bei dieser wird ein einzelner Radiofrequenzimpuls (RF-Puls) oder eine Sequenz von RF-Pulsen auf eine Probe gesandt, die sich in einem starken Magnetfeld befindet. Nach dem Abklingen des Pulses in der Empfangselektronik (Totzeit) wird der Zerfall der Magnetisierung (engl. free induction decay, FID), d. h., ihre Rückkehr in den Gleichgewichtszustand, über die dadurch in der Empfangsspule induzierte Spannung als Funktion der Zeit detektiert. Durch Fourier-Transformation wird dieses Zeitsignal im Computer in das Frequenzspektrum (Signalintensität als Funktion der Frequenz) transformiert.
Diese Messtechnik hat das früher verwendete CW-Verfahren (engl. continous wave) (s. o.) fast vollständig verdrängt.
Experimentelle Größen Bearbeiten
- Die chemische Verschiebung einer Resonanz ist vom lokalen Magnetfeld am Kernort abhängig, das wiederum von der chemischen Umgebung des betrachteten Kerns abhängt.
- Die Intensität einer Resonanz ist zumeist proportional zur Anzahl der sie hervorrufenden Kerne, solange die Besetzungsunterschiede nicht durch starke Kopplung oder Suszebtibilitätsunterschiede verändert werden.
- Bei den Relaxationszeiten angeregter Zustände unterscheidet man zwischen longitudinaler Relaxationszeit (Spin-Gitter-Relaxation) und transversaler Relaxationszeit (Spin-Spin Relaxation). Longitudinale Relaxationszeiten bestimmen die Einstellung der Gleichgewichtsmagnetisierung. Die transversalen Relaxationszeiten bestimmen die Linienbreite der Resonanzlinien. Relaxationseffekte geben Aufschluss über vorhandene Wechselwirkungen und molekulare Bewegungen.
- Räumlich benachbarte Kerne wechselwirken miteinander über magnetische Dipol-Dipol-Wechselwirkung (dipolare Kopplung). Diese Wechselwirkung verschwindet in isotropen Lösungen im zeitlichen Mittel.
- Indirekt können Kerne auch über chemische Bindungen miteinander wechselwirken. Diese skalare Kopplung ist für die Aufspaltung der Signale in Multipletts verantwortlich und stellt eine wesentliche Grundlage für die molekulare Strukturbestimmung mit NMR dar. Der Abstand zweier benachbarter Linien eines Multipletts wird als Kopplungskonstante, die in Hertz gemessen wird, bezeichnet.
Eindimensionale NMR-Spektroskopie Bearbeiten

Die eindimensionale NMR-Spektroskopie ist die am häufigsten angewandte Strukturaufklärungsmethode der Chemie. Bei ihr wird die chemische Verschiebung eines Atoms von einer Referenzsubstanz gemessen. 1H und 13C sind die Kerne, die am häufigsten in der organischen Chemie gemessen werden, aber auch 15N, 31P, 19F und viele andere NMR-aktive Isotope können spektroskopiert werden.

Das Aussehen der Spektren hängt entscheidend von der Aufnahmeart ab. 1H-Spektren werden in der Regel nicht Breitband-entkoppelt aufgenommen. Damit haben alle Wasserstoffatome die Möglichkeit, ihren Spin mit anderen Kernen zu koppeln, die sogenannte Spin-Spin-Kopplung. Damit entsteht bei der charakteristischen chemischen Verschiebung eines Atoms eine für seine Umgebung charakteristische Aufspaltung des Signals, aus der Informationen über die Molekülstruktur abgeleitet werden können.
13C, 15N, 31P, 19F und andere Kerne werden häufig 1H-Breitband-entkoppelt aufgenommen, so dass die Aufspaltung der Signale aufgrund der Kopplungen zu 1H-Kernen ausbleibt.
skalare Kopplung Bearbeiten
Der Kern eines Atoms kann mit einem benachbarten Atomkern in Wechselwirkung treten. Das kann entweder direkt (durch den Raum) oder indirekt (über die Bindungselektronen zwischen den Kernen) geschehen. Bei einer flüssigen Probe mittelt sich die direkte (dipolare) Wechselwirkung durch die schnelle Bewegung der Kerne im Raum aus. Erhalten bleibt die skalare Kopplung, die dadurch vermittelt wird, daß die (stets gepaart (↓↑)) auftretenden Spins der Bindungselektronen unterschiedlich mit den Kernspins auf beiden Seiten der Bindung wechselwirken.
Hat ein Kern den Zustand α (↑), so wird das (↑)-Elektron der Bindung von ihm abgestoßen, hält sich also eher am anderen Kern auf. Weitere von dort ausgehende Bindungen werden gleichfalls (in geringerem Ausmaß) Spinpolarisiert. Wird so ein weiterer Kern mit I>0 erreicht, ergibt sich ein Energieunterschied zwischen dessen Zustand α und β durch wiederum seine Wechselwirkung mit den Elektronen der Bindung. Solche Kopplungen sind gewöhnlich über max. drei bis vier Bindungen nachweisbar, in konjugierten π-Systemen z.B. aber auch weiter.
Das NMR-Signal des ersten Kerns wird dadurch zu einem Dublett aufgespalten, und das des zweiten Kerns gleichfalls, und zwar um den gleichen Betrag, da der Energieunterschied (sog. Kopplungskonstante) zwischen αα(=ββ) und αβ(=βα) derselbe sein muß. Durch die gleichstarke Aufspaltung ist dann die Nachbarschaft der beiden Atome im Molekül nachgewiesen. Koppelt ein Kern zu zwei (oder allgemein n) gleichartigen (mit gleicher chemischer Umgebung und Spin-Symmetrie), so erhält man ein Triplett (oder Quartett, Quintett, usw., also (n+1) Linien, bzw. Linien für Kerne mit I>1/2), deren relative Intensitäten sich (für I=1/2 - Kerne) aus der (n+1)-ten Zeile des Pascalschens Dreiecks ergeben, also 1:2:1 oder 1:3:3:1. Sind die Kopplungskonstanten unterschiedlich, so fallen die mittleren Linien nicht zusammen, man erhält dann z.B. ein Dublett vom Triplett o.ä.

Beispielspektren Bearbeiten
Als ein einfaches Beispiel dient Propan (H3C–CH2–CH3): Die CH2-Gruppe beim Propan hat zwei benachbarte Methylgruppen (–CH3). Dies entspricht sechs benachbarten, äquivalenten H-Atomen. Das Signal wird also in ein Septett aufgespalten. Die Methyl-Protonen werden von den beiden Methylen-Protonen zum Triplett aufgespalten, die 4J-Kopplung zu den drei anderen Methyl-Protonen ist zu schwach, um noch aufgelöst zu werden.
Sind in einem Molekül mehrere unterschiedliche z. B. Methyl-Gruppen vorhanden, so überlagernd sich deren Multipletts häufig, wodurch sie schnell unauswertbar werden. Um solche Fälle besser auflösen zu können, wird hierfür vielfach auf mehrdimensionale NMR-Techniken wie COSY zurückgegriffen. Da die Aufspaltung nicht feldabhängig ist, der Abstand zwischen den Signalen chemisch unterschiedlicher Protonen aber schon, können Überlagerungen auch durch Anwendung eines höheren Feldes aufgelöst werden.
Erklärungen zum Spektrum von Ethanol:
Die OH-Gruppe bildet nur ein Singulett, wenn das Ethanol in wässriger Lösung vorliegt. Das alkoholische Wasserstoffatom ist leicht acid, und wird deswegen ständig durch Wasserstoffatome aus dem Lösungsmittel ausgetauscht. Das führt dazu, dass keine permanenten Spin-Spin-Kopplungseffekte auftreten. In reinem Ethanol würde dort, wie wegen der CH2-Gruppe erwartet ( ), ein Triplett entstehen.

Beispiele zweidimensionaler NMR-Spektroskopie Bearbeiten
- COSY (engl. correlation spectroscopy)
- Zweidimensionale Methode, bei der gleichartige Kerne (1H) (homonukleare COSY) oder verschiedenartige Kerne (heteronukleare COSY) über ihre skalaren Kopplungen miteinander korreliert werden. COSY-Spektren sind symmetrisch bezüglich der Diagonalen. Mit COSY können komplizierte Kopplungsmuster räumlich entzerrt werden.
- DOSY (engl. diffusion ordered spectroscopy)
- Verfahren, bei dem mittels Feldgradienten-NMR Moleküle mit unterschiedlichem Diffusionsverhalten NMR-spektroskopisch getrennt erfasst werden können.
- TOCSY (engl. total correlated spectroscopy)
- Zweidimensionale Methode, bei der gleichartige Kerne (1H) über ihre skalaren Kopplungen miteinander korreliert werden. TOCSY-Spektren sind wie COSY-Spektren symmetrisch bezüglich der Diagonalen. Zusätzlich zu den im COSY detektierten Signalen erscheinen im TOCSY auch Korrelationen zwischen dem Startkern und sämtlichen indirekt über mehrere Kopplungen mit ihm verbundenen Kernen (Spinsystem). Das TOCSY-Experiment ist vor allem bei der Strukturaufklärung hochmolekularer Substanzen mit räumlich begrenzten Spinsystemen, wie etwa Polysacchariden oder Peptiden, sehr nützlich.
- HSQC (engl. heteronuclear single quantum coherence)
- Zweidimensionale Methode, bei der chemische Verschiebungen unterschiedlicher miteinander skalar koppelnder Nuklide korreliert werden. Die HSQC-Spektren sind häufig recht übersichtlich, da gewöhnlich nur Signale von direkt aneinander gebundenen Atomen erscheinen. Typische Beispiele sind 1H,13C- und 1H,15N-Korrelationen.
- HMBC (engl. heteronuclear multiple bond coherence)
- Zweidimensionale Methode, bei der chemische Verschiebungen unterschiedlicher miteinander skalar koppelnder Nuklide korreliert werden. Im Gegensatz zum HSQC werden im HMBC Korrelationen über mehrere Bindungen angezeigt. Typisch sind vor allem 1H,13C-Korrelationen.
- NOESY (engl. nuclear overhauser enhancement spectroscopy)
- Zweidimensionale Methode, mit der Korrelationen über den Kern-Overhauser-Effekt (NOE) anstatt über skalare Kopplungen detektiert werden. Mit dieser Methode können räumlich benachbarte Kerne erkannt werden, auch wenn sie nicht skalar miteinander koppeln. Es gibt sowohl homo- als auch heteronukleare Versionen. Dieses Verfahren wird häufig in der Strukturaufklärung eingesetzt.
All diese Varianten beruhen darauf, eine lange Reihe an Spektren aufzunehmen, und währenddessen einen Pulsparameter kontinuierlich in seiner Dauer zu verändern, wodurch sich Phase und Intensität der Spektren systematisch ändern. Durch erneute Fourier-Transformation der einzelnen Punkte der Spektren entlang dieser Zeitachse wird aus den eindimensionalen Spektren ein zweidimensionales erhalten. Durch Variation weiterer Parameter können auch höherdimensionale Spektren erhalten werden. Die benötigte Spektrenzahl und damit Meßzeit steigt dabei exponentiell an.
Niederfeld-NMR Bearbeiten
Es gibt relativ preiswerte Niedrigfeld-NMR-Geräte (≈ 10–40 MHz), die, mit einem Permanentmagneten ausgestattet, zwar keine aufgelösten Spektren liefern, dafür aber in den Betriebskosten unvergleichlich günstiger (keine He-Kühlung) sind. Auch können solche Systeme portabel ausgelegt werden. Durch Analyse der 1H-Relaxationszeiten können Mischungsanteile von Mehrstoffsystemen (Suspensionen, teilkristalline Substanzen[31]) und, nach Kalibrierung, auch Absolutmengen von Stoffen quantifiziert werden, was besonders in der Industrie interessant ist. Messungen erfolgen dabei anstatt in einem (teuren) deuterierten Lösungsmittel üblicherweise in Substanz. Andere Kerne als Wasserstoff werden aufgrund der geringen Empfindlichkeit nur selten untersucht.
Einzelnachweise Bearbeiten
- ↑ Frequenzkamm in astronomischen Beobachtungen
- ↑ Eintrag zu mass spectroscopy. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.M03748 – Version: 2.2.
- ↑ Eintrag zu mass spectrometry. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.M03746 – Version: 2.2.
- ↑ William Prout: On the relation between the specific gravities of bodies in their gaseous state and the weights of their atoms. In: Annals of Philosophy. 6, 1816, S. 321–330 (Online).
- ↑ William Prout: Correction of a mistake in the essay on the relation between the specific gravities of bodies in their gaseous state and the weights of their atoms. In: Annals of Philosophy. 7, 1816, S. 111–113 (Online).
- ↑ E. Goldstein: Canalstrahlen. In: Sitzungsbericht der Preussischen Akademie der Wissenschaften. Band 691, 1886, S. 691–699.
- ↑ E. Rückardt: Zur Erinnerung an Wilhelm Wien bei der 25. Wiederkehr seines Todestages. In: Die Naturwissenschaften. 42. Jahrgang, Heft 3, 1955, S. 57–62 doi:10.1007/BF00589524.
- ↑ E. Rückhardt: Zur Entdeckung der Kanalstrahlen vor fünfzig Jahren. In: Die Naturwissenschaften. 24. Jahrgang, Heft 30, 1936, S. 465–467, doi:10.1007/BF01473963.
- ↑ J. J. Thomson: Cathode Rays. In: Philosophical Magazine. 44, 1897, S. 293, doi:10.1080/14786431003659214 (facsimile von Stephen Wright, Classical Scientific Papers, Physics, 1964).
- ↑ J. J. Thomson: Rays of positive electricity. In: Proceeding of the Royal Society A. 89, 1913, S. 1–20 (Digitalisat auf JSTOR, wie exzerpiert in Henry A. Boorse, Lloyd Motz: The World of the Atom. Vol 1, 1966 PDF).
- ↑ F. W. Aston: Kanalstrahlen und Atomphysik. In: Die Naturwissenschaften. 24. Jahrgang, Heft 30, 1936, S. 467–469, doi:10.1007/BF01473964.
- ↑ Gohlke, R. S., Time-of-flight mass spectrometry and gas-liquid partition chromatography. Anal. Chem. 1959, 31, 535-41
- ↑ Gohlke, R. S.; McLafferty, F. W., Early gas chromatography/mass spectrometry. J. Am. Soc. Mass Spectrom. 1993, 4, (5), 367-371.
- ↑ M.B. Comisarow and A.G. Marshall, Chem. Phys. Lett. 25, 282 (1974)
- ↑ Munson, M.S.B.; Field, F.H. J. Am. Chem. Soc. 1966, 88, 2621-2630. Chemical Ionization Mass Spectrometry. I. General Introduction.
- ↑ Beckey H.D. Field ionization mass spectrometry. Research/Development, 1969, 20(11), 26
- ↑ Teach/Me Instrumentelle Analytik. Abgerufen am 9. Dezember 2010.
- ↑ A. M. Boehm et al.: Extractor for ESI Quadrupole TOF Tandem MS Data Enabled for High Throughput Batch Processing. In: BMC Bioinformatics. 5. Jahrgang, 162, 2004, doi:10.1186/1471-2105-5-162.
- ↑ A. M. Böhm: Methoden zur effizienten Proteinidentifizierung anhand von Massenspektrometrie. Logos-Verlag, 2006
- ↑ Die spinen, die Teilchen bei desy.de.
- ↑ Isidor Isaac Rabi (1898-1988), bei National High Magnetic Field Laboratory
- ↑ What is EPR ? bei ETH Zürch.
- ↑ Nobelpreisvortrag von F. Bloch, 1952.
- ↑ Continuous-Wave Nuclear Magnetic Resonance (NMR) Spectroscopy bei Department of Chemistry, University of Adelaide.
- ↑ Nicole Kresge, Robert D. Simoni, Robert L. Hill: Succeeding in Science Despite the Odds; Studying Metabolism with NMR by Mildred Cohn. In: Journal of Biological Chemistry. Band 279, Nr. 53, 31. Dezember 2004, S. e12, PMID 15615732 (Online [abgerufen am 18. August 2010]).
- ↑ Nobelpreisvortrag von Ernst.
- ↑ E. R. Andrew, Nuclear Magnetic Resonance, Cambridge at the University Press, 1958, S. 62.
- ↑ C. Reinhardt, T. Steinhauser: Formierung einer wissenschaftlich-technischen Gemeinschaft. NMR-Spektroskopie in der Bundesrepublik Deutschland. In: NTM Zeitschrift für Geschichte der Wissenschaften, Technik und Medizin. 16, 2008, S. 73–101, doi:10.1007/s00048-007-0280-z.
- ↑ Nobelpreisvortrag von Wüthrich.
- ↑ P. T. Callaghan: Principles of Nuclear Magnetic Resonance Microscopy. Clarendon Press, Oxford 1991.
- ↑ Andreas Maus, Christopher Hertlein, Kay Saalwächter: A Robust Proton NMR Method to Investigate Hard/Soft Ratios, Crystallinity, and Component Mobility in Polymers In: Macromolecular Chemistry and Physics. 207, 2006, S. 1150, doi:10.1002/macp.200600169.
Literatur Bearbeiten
- Malcom H. Lewitt: Spin Dynamics. Wiley & Sons, Chichester 2001, ISBN 0-471-48922-0.
- Harald Günther: NMR-Spektroskopie. 3. Auflage. Thieme, Stuttgart 1992, ISBN 3-13-487503-9.
- Ullmanns Enzyklopädie der Technischen Chemie. Band 5. 4. Auflage. Weinheim 1980, ISBN 3-527-20005-3, S. 382 ff.
- A. Streitwieser Jr., C. H. Heathcock: Organische Chemie. Verlag Chemie, Basel 1980, ISBN 3-527-25810-8, S. 205–249.
- Dudley H. Williams, Ian Fleming: Spektroskopische Methoden zur Strukturaufklärung, Kernmagnetische Resonanz-Spektren. Thieme, Stuttgart 1975, ISBN 3-13-437203-7, S. 80–161.
- H. Friebolin: Ein- und Zweidimensionale NMR-Spektroskopie. 4. Auflage, Wiley-VCH, Weinheim 2006 ISBN 3-527-31571-3.
- Siegmar Braun, Hans-Otto Kalinowski und Stefan Berger: 150 and More Basic NMR Experiments. A Practical Course. Wiley-VCH, Weinheim 1998, ISBN 3-527-29512-7.
Weblinks Bearbeiten
- Leicht verständliche Shockwave-Animation des NMR-Prinzips - University of Florida
- Verständliche Animation von BIGS zu Details des MRT, wie Pulssequenzen, Relaxieren oder Spinmodifizierung
- Verständliche Einführung in die Thematik von der TU-Berlin
- Einführung in die 1H-NMR-Spektroskopie
- Spektrendatenbank (nicht nur für NMR) (englisch)
- Kernspinresonanz – Einführung in die Theorie. Enthält viel Werbung und tote Links.
- Basics of NMR von Joseph P. Hornak, Ph. D. (englisch)
- e-MRI –Magnetic Resonance Imaging physics and technique course, von Campus Medica
- 1H-NMR Prediction - Onlineberechnung von 1H NMR Spectren (englisch)
- 1H-NMR ohne Formeln - Einführung für den Schulunterricht
- NMR-Parallel-Schwingkreise - Parallelschwingkreise für NMR-Messungen (PDF-Datei; 340 kB)
- James Shoolery, A Basic Guide to NMR, 3rd Edition, Stan's Library, Vol.II, 2008, DOI 10.3247/sl2nmr08.012 (PDF-Datei; 4,15 MB; englisch)
- nmr.ch - Swiss NMR Community (englisch)
Chromatographie Bearbeiten
Chromatographie, Chromatografie (griechisch, zu deutsch Farbenschreiben) wird in der Chemie ein Verfahren genannt, das die Auftrennung eines Stoffgemisches durch unterschiedliche Verteilung seiner Einzelbestandteile zwischen einer stationären und einer mobilen Phase erlaubt. Dieses Prinzip wurde erstmals 1901 von dem russischen Botaniker Michail Semjonowitsch Tswett beschrieben, 1903 wurde es zum ersten Mal öffentlich gedruckt beschrieben, 1906 benutzte er erstmals den Begriff „Chromatographie“. Er untersuchte gefärbte pflanzliche Extrakte, zum Beispiel aus Blattmaterial, und konnte daraus durch Chromatographie verschiedene Farbstoffe isolieren. Praktische Anwendung findet diese Methode zum einen in der Produktion zur Isolierung oder Reinigung von Substanzen (= präparative Chromatographie), zum anderen in der chemischen Analytik, um Stoffgemische in möglichst einheitliche Inhaltsstoffe zwecks Identifizierung oder mengenmäßiger Bestimmung aufzutrennen. Die Chromatographie wird in der organischen Chemie, der Biochemie, der Biotechnologie, der Mikrobiologie, der Lebensmittelchemie, der Umweltchemie und auch in der anorganischen Chemie angewendet.
Prinzip der Chromatographie Bearbeiten
Die Chromatographie lässt sich am einfachsten durch einen Vergleich erklären:
Ein reißender Fluss kann einiges an Treibgut mit sich führen. Die Geschwindigkeit, mit der das Treibgut weiterbewegt wird, hängt ab
- von der Art des Treibguts (Sandkörner werden schneller als Kieselsteine transportiert),
- von der Beschaffenheit des Flussbetts (raue Oberflächen erhöhen die Reibung des Treibguts und verringern somit die Geschwindigkeit des Abtransports)
- von der Strömungsgeschwindigkeit.
In der Chromatographie werden unterschiedliche Substanzen (= Treibgut) in der so genannten Mobilen Phase (= Wasser) auf einer Stationären Phase (= Flussbett) befördert. Aufgrund der Wechselwirkungen (siehe die Einteilung unter Trennprinzipien) zwischen der Probe, der stationären Phase und der mobilen Phase werden einzelne Substanzen unterschiedlich schnell weitertransportiert und werden somit voneinander getrennt: Ein Gemisch aus Sand, sehr kleinen und etwas größeren Kieselsteinen wird an einer Stelle des Flusses eingebracht; nach beispielsweise hundert Metern kommt zuerst der gesamte Sand an (verteilt auf ein paar Meter) und nach einer gewissen Wartezeit kommen alle kleineren Kieselsteine und noch viel später die größeren, jeweils auf eine gewisse Strecke auseinandergezogen.
Dieser Vergleich eignet sich für einen ersten Einstieg. Tatsächlich erinnert der Prozess (bei der Chromatographie) eher an einen „digitalen“ Prozess (stop and go). Die Probemoleküle werden entweder mit der mobilen Phase mitgenommen (mit der Geschwindigkeit der mobilen Phase – Analogie wäre ein Floß, das passiv in einem Strom mitgeführt wird) oder sie haften an der stationären Phase (Geschwindigkeit gleich Null). Zwischen diesen beiden Möglichkeiten wechseln sie sehr rasch hin und her (aufgrund der Wärmebewegung erhalten sie ständig Stöße). Der Vergleich mit dem Flussbett könnte auch zu einem weiteren Missverständnis führen: die Verzögerungen, die die verschiedenen Probemoleküle auf dem Weg durch das chromatographische System erleiden, haben nichts mit Reibungsphänomenen zu tun. Basis für das Verständnis sind Unterschiede in der Verteilung (der verschiedenen Molekülsorten A, B, C usw.). Sie entsprechen Unterschieden im Zeitanteil (den die einzelnen Moleküle vom Typ A, B, C usw. im Mittel in der mobilen Phase verbringen). Die Chromatographie schafft es, diese Unterschiede in Geschwindigkeitsunterschiede zu verwandeln und damit für eine Trennung gut nutzbar zu machen. Das könnte man auch als den „Trick“ oder als das Prinzip der Chromatographie bezeichnen. Ansonsten wären diese oft recht kleinen Unterschiede kaum zu nutzen, weder für Trenn- und Reinigungsprozesse noch für Analysen.
Anhand eines Beispiels ist es leichter zu verstehen: Wenn sich 45 % der A-Moleküle in der mobilen Phase aufhalten (im Mittel), kommt es auf Grund des dynamischen Gleichgewichtes dazu, dass die individuellen A-Moleküle auch 45 % der Zeit in der mobilen Phase verbringen (im Mittel). Daher wird ihre Geschwindigkeit 45 % der Geschwindigkeit der mobilen Phase betragen (im Mittel). Für gute Ergebnisse bei der Chromatographie ist es entscheidend, dass der Stoffaustausch zwischen den beiden Phasen sehr rasch erfolgt, das heißt die einzelnen Probemoleküle sollen sehr oft zwischen den beiden Phasen hin und her wechseln (Diffusionsprozesse, Wärmebewegung). Eine Voraussetzung dafür ist, dass die Wege, welche die Moleküle von der stationären Phase zur mobilen Phase zurückzulegen haben, sehr kurz sind. Falls die stationäre Phase ein Pulver enthält, sollte die Korngröße dieses Pulvers sehr klein sein (zum Beispiel nur wenige Mikrometer). Aus bestimmten Gründen sollten die Pulverkörner auch möglichst einheitlich geformt sein und eine möglichst einheitliche Größe aufweisen (enge Korngrößenverteilung).
Prozess Bearbeiten

Für die Chromatographie sind die Herstellung des Flusses der mobilen Phase, die Injektion der zu trennenden Probe, die eigentliche Trennung und die Detektion nötig. Das Fließen der mobilen Phase wird entweder mittels Druck (hydraulischen Pumpe, Gasdruck), Kapillarkraft oder durch Anlegen einer elektrischen Spannung erreicht.
Die Injektion (= Einbringen des Substanzgemisches in das chromatographische System) erfolgt entweder, bevor der Fluss der Mobilen Phase hergestellt wird (z. B. Dünnschichtchromatographie) oder während die Mobile Phase bereits fließt. Bei einer großen Anzahl von Proben werden bei automatisierbaren Chromatographiearten sogenannte Autosampler (zusammen mit eigenen Datenerfassungssystemen) eingesetzt, die vollautomatisch die Proben injizieren.
Anschließend erfolgt die eigentliche Auftrennung des Substanzgemisches auf der Trennstrecke. Ohne Detektion (= sichtbar machen, wann eine Substanz einen bestimmten Teil des Chromatographiesystems passiert oder wo eine Substanz nach dem Beenden des Prozesses zum Liegen kommt) ist eine Chromatographie nicht denkbar. Für jede Chromatographieart werden verschiedene Detektionsysteme eingesetzt, indem entweder physikalische Eigenschaften (Absorption von Licht, Fluoreszenz, Lichtstreuung, Wärmeleitfähigkeit.) der Substanzen ausgenutzt werden oder durch chemische Reaktionen ein Signal erhalten wird. Mittels chemischer Reaktionen wird z. B. eine Färbung bei der planaren Chromatographie erreicht (z. B. Aminosäuren mittels Ninhydrin) oder Reaktionen vor dem Auftrennen (Vorsäulenderivatisierung) oder nach dem Auftrennen (Nachsäulenderivatisierung) bei der Säulenchromatographie durchgeführt.
Bei der präparativen Chromatographie wird anschließend noch ein Fraktionensammler zum Auffangen der aufgetrennten Substanz benötigt.
Bauartbedingt handelt es sich bei chromatographischen Aufreinigungsverfahren immer um Batch-Verfahren. Dies bedeutet, dass immer nur eine bestimmte Menge an Substanz aufgetragen und getrennt werden kann, bevor mit der nächsten Menge fortgefahren werden kann. Dies ist insbesondere bei der Aufarbeitung großer Mengen problematisch, so dass einige Verfahren entwickelt wurden, um Chromatographie kontinuierlich betreiben zu können: Annulare Chromatographie, TMB (True Moving Bed) Chromatographie und SMB-(Simulated Moving Bed) Chromatographie.
Begrifflichkeiten und Prinzip Bearbeiten
Stationäre Phase Bearbeiten
Phase, die mit den einzelnen Substanzen des Substanzgemisches Wechselwirkungen eingeht und sich nicht bewegt. Der Aufenthalt der Analyten bei ihrer Retention wechselt zwischen mobiler und stationärer Phase (random walk), und verursacht die substanzcharakteristische Retentionszeit. Die stationäre Phase besteht in der Gaschromatographie aus einer Flüssigkeit (Trennflüssigkeit) oder einem Gel, mit welchem die Innenseite der Kapillare beschichtet ist. Bei der Flüssigchromatographie ist die stationäre Phase normalerweise fest.
Mobile Phase Bearbeiten
Phase, in die das Substanzgemisch am Beginn des Trennsystems eingebracht und die bewegt wird. Bei der Flüssigchromatographie ist die mobile Phase flüssig. Bei der Gaschromatographie kommen Trägergase wie Wasserstoff, Helium oder Stickstoff zum Einsatz, in der Dünnschichtchromatographie spricht man vom Fließmittel. Mobile Phasen unterscheiden sich in ihrer Elutionsfähigkeit („Stärke“ s. u. „Elutrope Reihe“), dies bedingt unterschiedliche Retentionszeiten und oft auch unterschiedliche Selektivitäten.
Retention Bearbeiten
Unter Retention versteht man den verzögerten Durchfluss von einzelnen Substanzen des Substanzgemisches der mobilen Phase durch Wechselwirkung mit der stationären Phase.
Die Retention einer Substanz durch die stationäre Phase wird im wesentlichen durch drei Aspekte bestimmt:
- Stärke der Wechselwirkung der Substanz mit der stationären Phase („Neigung in der stationären Phase zu bleiben“)
- Siedepunkt der Substanz („Neigung in der mobilen Phase zu bleiben“)
- Diffusionseigenschaften der Substanz („Beweglichkeit in der stationären und mobilen Phase“)
In vielen Fällen wird gezielt eine spezielle Wechselwirkung des zu analysierenden Stoffes mit der stationären Phase genutzt, um Substanzen zu trennen. Die Stärke der Wechselwirkungen zwischen den Probenkomponenten und der stationären Phase wird sowohl von deren Struktur als auch von deren funktionellen Gruppen bestimmt. Dabei treten bei unpolaren Substanzen ausschließlich Dispersionswechselwirkungen (Van-der-Waals-Bindung) auf, während polare Trennphasen auch polare Wechselwirkungen eingehen können, etwa Wasserstoffbrückenbindungen oder Donator-Akzeptor-Bindungen. Letztere trennen nach dem Prinzip: Gegensätze ziehen sich an. Das bedeutet, dass Trennphasen, die etwa Wasserstoff zur Wasserstoffbrückenbindung aufzunehmen in der Lage sind Substanzen trennen, die Wasserstoff zur Brückenbindung bereitstellen können (etwa Alkohole). Auch können zum Beispiel Enantiomere, welche sich in ihren Siedepunkten nicht unterscheiden und somit gleiche Retentionszeiten aufweisen würden, durch ihre verschieden starken Wechselwirkungen mit speziellen Derivaten von Cyclodextrinen aufgetrennt werden.
Retentionszeit Bearbeiten
Zeit, die die Moleküle eines reinen Stoffes zum Durchwandern der Säule benötigen (von der Injektion bis zur Detektion).
Im Gegensatz zu den Trägergasen zeigen die meisten chemischen Stoffe eine Wechselwirkung mit der stationären Phase, d. h. sie halten sich für eine gewisse Zeit in der stationären Phase auf. Ihre Aufenthaltsdauer in der stationären Phase addiert sich zur Aufenthaltsdauer in der mobilen Phase (Totzeit), sie benötigen also insgesamt länger, um die ganze GC-Säule zu passieren. Ursprünglich leitet sich der Begriff Retention (Zurückhaltung) davon ab, dass die stationäre Phase den Analyten für eine gewisse Zeit zurückhält. Heutzutage wird der Begriff Retentionszeit allerdings vereinfacht verwendet für die Zeit, die der Analyt zum Passieren der Säule benötigt und dies schließt also die Totzeit mit ein. Daher werden die Begriffe folgendermaßen definiert:
- Retentionszeit [tR]: Ist die Gesamtzeit, die ein Analyt für das Passieren der Säule benötigt. Dies entspricht der Zeit zwischen Injektion und Detektion des Analyten.
- Totzeit [t0]: Ist die Zeit, in der sich ein Analyt ohne Wechselwirkung mit der stationären Phase in der mobilen Phase aufhält; entspricht also der Zeit, die die mobile Phase zum Durchlaufen der Säule benötigt.
- Nettoretentionszeit oder reduzierte Retentionszeit [tN]: Ist die Differenz aus Retentionszeit und Totzeit. Sie entspricht also der Zeit, in der sich ein Analyt in der stationären Phase aufhält. tN = tR - t0
Durchflusszeit (Totzeit) Bearbeiten
Die Durchflusszeit (früher auch „Totzeit“) gibt die Zeit an, die die mobile Phase oder eine nicht zurückgehaltene Substanz benötigt, um die Chromatographie-Apparatur von der Injektion über die Säule bis zum Detektor zu durchwandern (s. a. Durchflussvolumen). Die Durchflusszeit kann bestimmt werden, indem eine nicht zurückgehaltene Substanz („Inertsubstanz“) in das HPLC-System injiziert wird. Diese Substanz geht nur in sehr geringem Maße Wechselwirkungen mit der stationären Phase ein. Sie durchläuft daher die Apparatur in derselben Zeit wie die mobile Phase. Die Durchflusszeit ist dann identisch mit der Zeit zu der der Peak im Detektor erscheint.
Durchflussvolumen Bearbeiten
Das Durchflussvolumen kann direkt aus der Durchflusszeit abgeleitet werden. Es ergibt sich aus der einfachen Formel Durchflussvolumen = Fluss der mobilen Phase • Durchflusszeit. Das Durchflussvolumen ist für zahlreiche Berechnungen in der HPLC sehr wichtig, z. B. für den Methodentransfer zwischen Säulen mit unterschiedlichem Volumen.
Elution Bearbeiten
Elution (von lat. eluere „auswaschen“) ist das Herauslösen oder Verdrängen von adsorbierten Stoffen aus festen oder mit Flüssigkeit getränkten Adsorbentien und Ionenaustauschern durch kontinuierliche Zugabe eines Lösungsmittels (Elutionsmittel = mobiler Phase). Die aus der Trennsäule fließende Lösung wird Eluat genannt.
Eine besondere Bedeutung hat dieser Prozess in der Festphasenextraktion.
Eluotrope Reihe Bearbeiten
Anordnung der als mobile Phase üblichen Lösungsmittel nach ihrer Elutionskraft bei einer Referenzsubstanz (i. d. R. Kieselgel oder Aluminiumoxid).
Bluten Bearbeiten
Als Bluten wird ein Effekt bei Chromatographie-Säulen bezeichnet, bei dem die Säule geringe Anteile ihrer Matrix verliert. Man spricht auch von Säulenbluten. Ursache für verstärktes Säulenbluten können in der Gaschromatographie eine übermäßige thermische Belastung der Säule und in der HPLC die Verwendung zB. ungeeigneter pH-Werte des Elutionsmittels oder der Eluenten (zu stark sauer oder alkalisch) sein. Säulenbluten findet in geringem Ausmaß auch im laufenden Betrieb statt und stellt dort normalerweise kein Problem dar. Durch das Säulenbluten ergibt sich, unter anderem, die Alterung von Trennsäulen. Starkes Säulenbluten bewirkt jedoch ein starkes Signalrauschen und einen hohen Hintergrundwert bei der Detektion oder nachgeschalteten Analyseverfahren, wie einem Massenspektrometer.
Säule Bearbeiten
In der Chromatographie versteht man unter einer Säule eine hohle Röhre mit einem Durchmesser von wenigen Mikrometern bis zu mehreren Metern. In dieser Röhre ist entweder nur die Innenwand beschichtet (Kapillarsäule) oder die Säule ist mit der stationären Phase befüllt (gepackte Säule).
Reversed-Phase-Mechanismus Bearbeiten
Bei der Adsorptionschromatographie gibt es zwei Möglichkeiten ein Substanzgemisch zu trennen:
- Normale Phase: polare stationäre Phase (wie Kieselgel, Aluminiumoxid), unpolare bis mittelpolare mobile Phase (wie Kohlenwasserstoffe, Dioxan, Essigsäureethylester …) oder
- Reversed Phase (Umkehrphase): unpolare stationäre Phase (wie modifiziertes Kieselgel) und polare mobile Phase (wie gepuffertes Wasser).
Im ersten Fall werden lipophile Stoffe leicht, polare schwer eluiert, im Umkehrfall polare leicht eluiert („similia similibus solvuntur“).
In der HPLC wird oft eine Gradienteneluierung angewandt (reversed phase), wobei die Zusammensetzung des Lösungsmittels langsam geändert wird (z. B. von 80 % auf 20 % Wasseranteil). So treten Alkane sehr spät und Aminosäuren sehr früh aus der Säule aus und man kann diese Fraktionen herausschneiden.
Einteilung nach dem Trennprinzip Bearbeiten
Das grundlegende Prinzip aller chromatographischen Verfahren ist die oft wiederholte Einstellung eines Gleichgewichtes zwischen einer ruhenden Phase und einer bewegten Phase. Das Gleichgewicht kann sich auf Grund verschiedener physikalisch-chemischer Effekte ausbilden.
- Adsorptionschromatographie – Hier kommt es zu einer Trennung der verschiedenen Komponenten auf Grund der unterschiedlich starken adsorptiven Bindungen zur ruhenden Phase. Die bewegte Phase kann ein mehr oder weniger polares Lösungsmittel oder bei gasförmigen Stoffen ein Trägergas sein. Im Fall der Flüssigchromatographie stellt man sich einen Wettkampf zwischen den diversen Probemolekülen mit den Molekülen der mobilen Phase (Fließmittel, Laufmittel) um die Haftstellen auf der (großen) Oberfläche der stationären Phase vor.
- Verteilungschromatographie – Ähnlich dem Extraktionsverfahren wird hier die unterschiedliche Löslichkeit der zu trennenden Komponenten ausgenutzt. Bei der Chromatographie bleibt aber das Lösungsmittel als ruhende Phase auf einem Trägermaterial haften. Die bewegte Phase kann wieder eine Lösung oder ein Trägergas sein.
- Ionenaustauschchromatographie – Die bewegte Phase ist hier meist eine Lösung der zu trennenden Ionen. Die ruhende Phase ist ein fester Ionenaustauscher. Ionenaustauscher bilden zwischen den verschiedenen Ionen der bewegten Phase unterschiedlich stabile Bindungen aus.
- Siebwirkung – Bei der ruhenden Phase benutzt man Stoffe, die die Komponenten anhand ihrer Größe trennen. Im Wesentlichen unterscheidet man hier zwischen drei Verfahren.
- Molekularsieb-Chromatographie
- Gel-Permeations-Chromatographie (Gelfiltration)
- Ausschluss-Chromatographie
- Bei einem Sieb haben Teilchen „Vorteile“, die fein genug sind, um die Poren des Siebes zu durchdringen. Bei den entsprechenden Chromatographieverfahren ist es genau umgekehrt. Genügend feine Teilchen sind in der Lage, sich in die Hohlräume der stationären Phase zu „verirren“, sind daher langsamer unterwegs als Moleküle, die auf Grund ihrer Größe aus diesen Hohlräumen (mehr oder weniger oder zur Gänze) ausgeschlossen werden. Bei genügender Größe wandern sie unverzögert, da sie sich nur im bewegten Flüssigkeitsstrom aufhalten und nie im unbewegten Teil der Flüssigkeit (in den Hohlräumen – z. B. des Gels).
- Affinitätschromatographie – Als stationäre Phase wird eine für jeden Analyten spezifische chemische Verbindung eingesetzt, die auf Grund nichtkovalenter Kräfte eine Trennung bewirkt. Es handelt sich hierbei um eine hochselektive Methode.
- IMAC (Immobilized Metal Ion Affinity Chromatography) – Hierbei werden über Komplexbindungen Metallionen wie Fe, Co, Ga u. a. an eine Matrix gebunden. Die Trennung wird über die unterschiedlichen Wechselwirkungen zwischen dem Analyten und den Metallionen erreicht. Besonders bei der Aufreinigung von Proteinen durch Phosphorylierung hat sich diese Methode etabliert. Dabei werden als Metallionen vornehmlich Fe und Ga genutzt. Als Konkurrenzverfahren hat sich dort seit einiger Zeit die Anreicherung über Titandioxid-Säulen bewährt. Für die Aufreinigung von poly-Histidin-markierten Proteinen sind ebenfalls verschiedene IMAC-Verfahren vorhanden. Hierbei werden vor allem Ni und Co Ionen für die Anreicherung genutzt.
- Chirale Chromatographie – Zur Trennung von chiralen Molekülen. Die stationäre Phase enthält ein Enantiomer, das mit den beiden Enantiomeren des Racemats eine unterschiedlich starke diastereomere Wechselwirkung eingeht. Die beiden Enantiomere werden unterschiedlich stark retardiert.
Einteilung nach den verwendeten Phasen Bearbeiten
Aufgrund der mobilen Phasen kann man die Chromatographie in drei Gebiete unterteilen, welche sich nach den Trägern der stationären Phasen oder dem Aggregatzustand der stationären Phasen wieder in unterschiedlich viele Gruppen unterteilen lassen.
- Flüssigchromatographie (engl. liquid Chromatography, LC)
- Planare Chromatographie
- Papierchromatographie – Als feste Phase wird Papier verwendet, das entweder liegt oder (meist) senkrecht in einem Glasbehälter steht. Wie auch bei der Dünnschichtchromatographie wird die mobile Phase auf Grund der Kapillarkräfte bewegt.
- Dünnschichtchromatographie – Als feste Phase werden z. B. Silikapartikel in einer feinen Schicht auf einer flexiblen Trägerfolie aus Aluminium oder Plastik oder einer Glasplatte aufgetragen. Eine Variante ist die zirkuläre DC, mit einer rotierenden, beschichteten Kreisscheibe (speziell für präparative Zwecke geeignet).
- Säulenchromatographie
- Niederdruckchromatographie – Die hier verwendeten Säulen weisen Durchmesser von einem bis mehreren Zentimetern auf. Diese Form der Flüssigchromatographie wird vor allem für präparative Trennungen eingesetzt.
- Hochleistungschromatographie (unrichtig, jedoch sehr gebräuchlich ist auch der Ausdruck Hochdruckchromatographie; engl. HPLC High Performance (Pressure) Liquid Chromatography) – Sie stellt die heute am weitesten verbreitete in der Analytik eingesetzte Trennmethode dar, die eigentlich unkorrekte (veraltete) Bezeichnung High Pressure Liquid Chromatography bezieht sich auf die Drücke, die diese Methode von der Niederdruck- oder anderen Chromatographiearten unterscheidet. Immerhin werden hier bis über 1000 bar bei einer Flussrate der mobilen Phase bis zu 5 ml/min erzeugt, die jedoch mit der Trennleistung nicht zu tun haben, sondern nur zur Fortbewegung des Eluentengemischs in der Säule dienen.
- Elektrochromatographie – In diesem Fall wird die Mobile Phase durch Anlegen einer Spannung bewegt. Diese Methode befindet sich noch im Entwicklungsstadium und wird im Routinebetrieb nicht angewendet. Nicht zu verwechseln mit Elektrophorese.
- Membranchromatographie
- Hierbei wird statt einer mit chromatographischer Matrix gefüllten Säule eine ein- oder mehrlagige Membran als feste Phase in einem entsprechenden Gehäuse eingesetzt. Die mobile Phase wird bei niedrigen Drücken bis zu 6 bar und bei etwa 20-fach höheren Flussraten als in der Säulenchromatographie üblich durch die Membran gepumpt.
- Planare Chromatographie
- Gaschromatographie
- Gepackte Säulen – Das Innere einer Säule (lange Röhre) ist mit einem feinkörnigen Material gefüllt. In der Regel besteht dabei die stationäre Phase aus einem dünnen Film einer weitgehend inerten und hochsiedenden Flüssigkeit, der die Pulverkörner überzieht.
- Kapillarsäulen – Nur die Säulenwand ist mit einer dünnen Schicht aus stationärer Phase bedeckt.
- Flüssige stationäre Phase
- Feste stationäre Phase
- Überkritische Fluidchromatographie (engl. SFC supercritical fluid chromatography) – Als mobile Phase wird eine Substanz in ihrer überkritischen Phase (Zustand zwischen Gas und Flüssigkeit) eingesetzt. Hierbei handelt es sich meist um Kohlendioxid. Bei dieser Methode werden nur Säulen als Träger von stationären Phasen eingesetzt.
Kenngrößen der Chromatographie Bearbeiten
- Säulenlänge L


- nennt man die lineare Durchflussgeschwindigkeit der mobilen Phase durch die Säule, sie ist definiert als:


- der Retentionsfaktor ist definiert durch


- Der Selektivitätskoeffizient α gibt die Güte der Trennung zweier Substanzen an. Er beruht auf der Retentionszeiten der Komponenten in der Säule. Die Retentionszeit ist die Zeit, die die betrachtete Komponente zum Durchqueren der Säule braucht und wird am Peakmaximum abgetragen:

- , die chromatographische Auflösung (Resolution) zweier Peaks errechnet sich aus:

- oder
- Der Faktor 1,18 kommt durch das Verhältnis der Halbwertsbreite zur Basisbreite einer Gaußschen Glockenkurve ((2 · ln 2)0,5) zustande.


- , die Trennstufenzahl oder Bodenzahl beschreibt die Anzahl der Gleichgewichtseinstellungen, der zu trennenden Substanz zwischen stationärer und mobiler Phase, in der Säule. Je größer N, desto mehr Gleichgewichtseinstellungen können in einer bestimmten Länge erfolgen, woraus eine bessere Trennleistung der Säule resultiert. N wird berechnet mit Hilfe der Formel:

- oder


- : Basislinienbreite

- : „Full Width at Half Maximum“ Fwhm

- : Peakkapazität; Gibt an, wie viele Peaks innerhalb eines Intervalls zwischen und dem k-Wert eines bestimmten Peaks theoretisch mit einer Auflösung von R=1,5 (Basislinientrennung) voneinander getrennt werden können.

- bezeichnet die Trennstufenhöhe (oder theoretische Bodenhöhe) eines theoretischen Bodens (HETP) und ist das Verhältnis zwischen Säulenlänge L und Bodenzahl N:


- Praktische Werte liegen im Bereich von 0,1 bis 0,5 mm.
Trennstufenhöhe H Bearbeiten
Die Trennstufenhöhe einer chromatographischen Säule ist ein Maß für die Trennleistung der Säule. Als Trennstufe kann man sich den gedachten Abschnitt der Trennsäule, auf dem sich das chromatographische Gleichgewicht einmal einstellt, vorstellen. Je mehr solche Gleichgewichtseinstellungen „auf der Säule Platz haben“ umso geringer ist die Trennstufenhöhe und umso höher ist die Trennleistung der Säule. Zur Erlangung einer niedrigen Trennstufenhöhe sind unter analytischen Bedingungen folgende Voraussetzungen nötig:
- Es wird eine rasche Gleichgewichtseinstellung der Adsorption oder Verteilung erwartet. Daher sollte der Teilchendurchmesser so klein wie möglich sein.
- Konstante Temperatur in der gesamten Säule. Dazu kann ein Säulenthermostat benutzt werden.
- Konstante Fließgeschwindigkeit: Hierfür wird eine Kolbenpumpe mit bis zu 400 bar verwendet.
- Linearer Adsorptionsbereich: Die stationäre Phase sollte im Verlauf der Chromatographie nicht überladen werden.
- Vernachlässigbare Diffusion wäre wünschenswert, ist experimentell aber leider nicht erreichbar. Es werden daher möglichst regelmäßige Packungen mit Teilchen von besonders kleinem Durchmesser verwendet.
Zur Ermittlung der Trennstufenhöhe in Abhängigkeit von der Fließgeschwindigkeit des Eluenten kann die sog. Van-Deemter-Gleichung für die HPLC herangezogen werden:

wobei:
- die Trennstufenhöhe,
- die lineare Fließgeschwindigkeit ist.
- -Term berücksichtigt die Eddy-Diffusion die durch unterschiedliche Fließstrecken durch die Packung entsteht. Es gilt: wobei
- den Packungsfaktor,
- den Teilchendurchmesser bezeichnet.
- Der -Term berücksichtigt die longitudinale Diffusion. Die longitudinale Diffusion ist die Diffusion der Analytenmoleküle in beide Richtungen der Trennstufe. Es gilt: wobei:
- die Diffusionskonstante in der mobilen Phase und
- der Labyrinthfaktor ist. Der Labyrinthfaktor berücksichtigt die Porenstruktur der stationären Phase.
- der -Term berücksichtigt die Peakverbreiterung durch die langsame Gleichgewichtseinstellung zwischen der mobilen und der stationären Phase. Hierbei ist noch die Diffusionskonstante entlang der Poren der stationären Phase zu beachten. Es gilt












Peaksymmetrie Bearbeiten
Theoretisch sollte jede Substanz eine Chromatographiesäule als scharf eluierende Linie verlassen. Aus verschiedenen Gründen besitzen chromatographische Peaks jedoch immer eine gewisse Breite. Im Idealfall weisen sie dabei die Form einer Gauß’schen Glockenkurve auf. In der Praxis kommt es aber häufig vor, dass die Peaks von dieser Idealform abweichen und mehr oder weniger asymmetrisch erscheinen. Eine Asymmetrie, bei der der Frontanstieg des Peaks steiler ist als der Peakabfall, bezeichnet man als „Tailing“, während der Effekt, dass der Anstieg weniger steil ist als der Abfall als „Fronting“ bezeichnet wird. Der Tailingfaktor, der ein Maß für die Peaksymmetrie darstellt, wird bestimmt, in dem man vom Peakmaximum das Lot zur Basislinie fällt, und in einer bestimmten Höhe, meist in 10 % der Peakhöhe, die Abstände zur Peakfront (a) und zum Peakende (b) ermittelt. Anschließend wird der Quotient der beiden Werte gebildet, wobei unterschiedliche Berechnungsformeln (z. B. nach IUPAC oder nach USP) in Gebrauch sind:

Ein idealer „Gauß-Peak“ erreicht dabei den Wert 1, Werte über 1 bedeuten „Tailing“, Werte unter 1 dagegen „Fronting“.
Verfahren Bearbeiten
- Anionenaustauschchromatographie
- Papierchromatographie
- Dünnschichtchromatographie
- Flüssigchromatographie
- Frontalchromatographie
- Gaschromatographie
- Gel-Permeations-Chromatographie
- Multidimensionale Chromatographie
Weblinks Bearbeiten
- Analytische Chemie – Chromatographie auf chemgapedia.de
- Einführung in die Chromatographie (englisch)
- Seite zu Chromatographie am NTK Landau
- Umfangreiche Übersicht von Herstellern für LC-Systeme
- Gute Beispielanwendungen zur Chromatographie in der Proteinanalytik
- Deutsche chromatographische Grundbegriffe zur IUPAC-Nomenklatur
Flüssigchromatographie Bearbeiten
Die Flüssigchromatographie, in der Standardsprache auch Flüssigchromatografie (engl.: liquid chromatography; abgekürzt LC) ist als Spezialgebiet der Chromatographie eine physikalische Trennmethode. Als Mobile Phase dient eine Flüssigkeit, als Stationäre Phase ein Feststoff oder eine Flüssigkeit.
Man unterscheidet:
- Papierchromatographie,
- Dünnschichtchromatographie (kurz DC) und
- Säulenchromatographie
- Feld-Fluss-Fraktionierung (kurz FFF)
Dabei unterscheidet man offene Systeme (z. B. Dünnschichtchromatographie, Papierchromatographie), wobei die stationäre Phase herausgenommen und direkt untersucht werden kann, und geschlossene Systeme (z. B. Säulenchromatographie, HPLC).
Bei geschlossenen Systemen werden die Substanzen nach Elution mit einem Lösungsmittel isoliert oder mit einem Detektor angezeigt. Ob diese Trennmethode zur Trennung eines Substanzgemisches eingesetzt werden kann, hängt vor allem davon ab, ob alle Substanzen des Gemisches in der Mobilen Phase gelöst werden können und ob es eine stationäre Phase gibt, die eine ausreichende Selektivität zwischen den Substanzen aufweist. Durch Elution oder Kapillarwirkung des Lösungsmittels kommt es durch die stationäre Phase zu einem Sortierprozess entsprechend den Moleküleigenschaften. Durch das nachlaufende Lösungsmittel bilden sich Substanzzonen aus, die am unteren Rohrausgang aufgefangen werden können.
Als stationäre Phasen kommen häufig modifizierte und nicht modifizierte Silikagele (auch Kieselgele genannt) und Polymere, z. B. Polystyrol-Divinylbenzol-Copolymere (PS/DVB), zum Einsatz. In der Feld-Fluss-Fraktionierung werden verschiedene Trennfelder (Flussfelder, thermische Felder, etc.) als stationäre Phase eingesetzt.
Papierchromatographie Bearbeiten

Die Papierchromatographie, in der Standardsprache auch Papierchromatografie, ist ein chromatographisches Trennverfahren für kleine Substanzmengen, bei dem ein feines Filtrierpapier die stationäre (ruhende) Phase und ein Lösungsmittel die mobile (bewegliche) Phase darstellt.[1]
Verfahren Bearbeiten

Die gelöste Probe wird in einem kleinen Tropfen auf das Filterpapier aufgebracht, daneben meist ein oder mehrere Tropfen mit einer oder mehreren bekannten Vergleichssubstanz(en). Das getrocknete Filterpapier wird so in ein geschlossenes Glasgefäß gestellt oder gehängt, dass das Papier nicht die Glaswand berührt. Der Startpunkt befindet sich am unteren Ende so weit vom Papierrand entfernt, dass er nicht in das Lösungsmittel eintaucht. Durch die Kapillarwirkung steigt das Lösungsmittel nach oben und transportiert die Substanzen mit, wobei sich durch die Wechselwirkung (Adsorption und Desorption) der Substanz mit dem Papier, genauer mit dem durch Luftfeuchtigkeit darauf enthaltenem Wasser, eine substanzspezifische Wanderungsgeschwindigkeit ergibt. Dieses Trennverfahren wird auch „aufsteigende“ Papierchromatographie genannt und ist weiter verbreitet als die sogn. „absteigende“ Papierchromatographie.[1]

Gebräuchlich ist neben der eindimensionalen Papierchromatographie auch die zweidimensionale Papierchromatographie (zweimaliger Durchlauf der Substanz bei Drehung des Chromatogramms um 90°) für die Trennung komplexerer Gemische, wie beispielsweise Aminosäuren aus Proteinen. Farblose Proben werden nach dem Trocknen des Chromatogramms mit einem Reagenz sichtbar gemacht, bei Aminosäuren beispielsweise durch Aufsprühen von Ninhydrin.
Die Papierchromatographie eignet sich sehr gut als Schüler-Experiment, da ohne Gefahr mit einfachen Mitteln ein wichtiges Analyseverfahren selbst durchgeführt werden kann. So kann z. B. die Farbstoff-Zusammensetzung verschiedener Filzstift-Tinten untersucht werden. Als Lösungsmittel wird Wasser verwendet. Neben dem Prinzip der Chromatographie werden hierbei auch grundlegende Erkenntnisse der subtraktiven Farbmischung vermittelt.
Preisverleihung Bearbeiten
Der Nobelpreis für Chemie des Jahres 1952 wurde an Archer John Porter Martin und Richard Laurence Millington Synge für die Entwicklung dieser Chromatographieart verliehen.
Literatur Bearbeiten
Friedrich Cramer: Papierchromatographie. Verlag Chemie, Weinheim 1953 und Neuauflagen.
= Dünnschichtchromatographie 0

Die Dünnschichtchromatographie (DC oder TLC, englisch thin layer chromatography) ist ein physikalisch-chemisches Trennverfahren, das zur Untersuchung der Zusammensetzung von Proben genutzt wird. Besonders vorteilhaft bei dieser chromatographischen Methode ist der geringe apparative Aufwand, die Schnelligkeit, die hohe Trennleistung und der geringe Substanzbedarf. Eingesetzt wird sie zum Beispiel zum raschen Nachweis der Reinheit einer Substanz oder der Überprüfung der Identität mit einer Referenzsubstanz. Auch die Verfolgung des Reaktionsverlaufes von chemischen Umsetzungen im Labor ist mit wenig Aufwand möglich.
Geschichte Bearbeiten
N. A. Izmailov und M. S. Shraiber, zwei russische Forscher, führten 1938 eine chromatographische Trennung mit einer horizontalen Dünnschichtplatte durch, auf die sie das Lösemittel auftropften. Doch ihre Methode wurde kaum beachtet. Erst als J. G. Kirchner und seine Mitarbeiter (darunter auch B. Harnischmacher) sich ab 1951 mit ihr befassten, wurde das Interesse anderer an der Methode geweckt. Zum Durchbruch verhalf ihr Egon Stahl, als er die Herstellung von leistungsfähigen Platten beschrieb. Von ihm stammt auch der Name Dünnschichtchromatographie.[2]
Theoretische Grundlagen Bearbeiten
Grundprinzip der chromatographischen Trennung Bearbeiten
Das Grundprinzip der Chromatographie gilt für alle chromatographischen Methoden und kann wie folgt kurz zusammengefasst werden: Teilchen (Moleküle, Ionen) verteilen sich auf zwei Phasen in einem bestimmten Verhältnis (Gleichgewichtszustand). Bei der DC wandert ein Lösungsmittel durch Kapillarkräfte an einem festen, feinporigen Trägermaterial (z.B. Kieselgel) aufwärts. Sind in dem Lösungsmittel unterschiedliche organische Substanzen mit verschiedenartigen funktionellen Gruppen enthalten, so gibt es Adsorptionswechselwirkungen der einzelnen Substanzen mit dem Trägermaterial. Eine starke Wechselwirkung dämpft die Wanderungsgeschwindigkeit der einzelnen Komponente in Bezug zur Lösungsmittelfront. Je höher der Anteil eines stark polaren Lösungsmittels (Wasser ist besonders schlecht) bei der Auftragung, desto mehr Oberflächenbereiche der Beschichtung sind für die Adsorption unwirksam, desto schlechter das Trennergebnis bei der DC (geänderte Rf-Werte, Fleckenverbreiterung).[3] Entscheidend ist, dass sie individuell sehr rasch von der einen Phase in die andere wandern (auf Grund der Wärmebewegung, der Diffusion und der raschen Austauschprozesse) und wieder zurück (dynamischer Gleichgewichtszustand). Der Zeitanteil, den das individuelle Teilchen in der mobilen bzw. der stationären Phase zubringt, entspricht auch genau dem Anteil der Teilchen dieser Sorte in den beiden Phasen. Diese Verhältnisse gelten auch, wenn die mobile Phase nicht bewegt wird.
In der Chromatographie werden die Unterschiede, die es bei den Austauschvorgängen zwischen den verschiedenen Teilchensorten gibt, in Geschwindigkeitsunterschiede verwandelt. Die Geschwindigkeit ergibt sich aus dem Produkt der Geschwindigkeit der mobilen Phase und dem Zeitanteil, den die Teilchen in der mobilen Phase verbringen. Es wird angenommen, dass die Teilchen in der mobilen Phase (statistisch gesehen) dieselbe Geschwindigkeit haben wie die Laufmittelmoleküle. Sind sie an die stationäre Phase gebunden, ist die Geschwindigkeit gleich Null („stop and go“-Modell). So werden also Verteilungsunterschiede (Verteilung im allgemeinen Sinn) in Geschwindigkeitsunterschiede verwandelt. Oft sind die Verteilungsunterschiede nur gering. Auftrennungen wären so mit anderen Methoden kaum zu erzielen. In dem Augenblick, in dem es sich aber um Geschwindigkeitsunterschiede handelt, ist es nur eine Frage der Länge der Wegstrecke, bis es zu einer ausreichenden Auftrennung gekommen ist.
Mit flüssigchromatographischen Methoden, zu denen auch die Dünnschichtchromatographie zählt, lassen sich alle Proben, die ausreichend stabil sind und in Lösung gebracht werden können, untersuchen.
Grundlage der Dünnschichtchromatographie sind also Transportprozesse in einer Flüssigkeit („mobile Phase“), die durch eine Pulverschicht („stationäre Phase“) strömt. Dabei zeigen unterschiedliche Moleküle meist unterschiedliches Wanderungsverhalten. Wie groß diese Unterschiede sind, ist abhängig von den chemischen und physikalischen Eigenschaften der verwendeten stationären und mobilen Phase.
Stationäre Phase Bearbeiten
In der Regel kommt als stationäre Phase Kieselgel zum Einsatz (Normalphasenchromatographie), das aufgrund der freien endständigen Hydroxygruppen als polares Adsorbens für die Probenmoleküle dient. Der mittlere Porendurchmesser der Kieselgele beträgt meist 4 bis 100 nm, wobei der Porendurchmesser von 6 nm (Kieselgel 60, Merck) am gebräuchlichsten ist. Kieselgele enthalten Siloxan- oder Silanol-Gruppen.
Alternativ dazu kommen auch DC-Materialien mit anderen funktionellen Gruppen (z. B. Aminogruppen) zum Einsatz. Sie unterscheiden sich vom Standard-Kieselgel nicht nur in ihrer Polarität, sondern auch in der Basizität und führen damit zu völlig anderen Trennergebnissen. Auch oberflächenmodifizierte Kieselgele mit unpolaren Haftstellen (durch Kupplung mit Organochlorsilanen) werden eingesetzt (Umkehrphasenchromatographie, reversed phase). Die Reihenfolge, in der die verschiedenen Probemoleküle aufgetrennt werden, kehrt sich dann um – die polaren Moleküle laufen schneller, die unpolaren Moleküle werden stärker festgehalten. Vorteilhaft ist dabei unter anderem, dass auch sehr polare Proben untersucht werden können. Zur Trennung chiraler Proben werden beispielsweise reversed phase-DC-Platten eingesetzt, die mit dem Kupfer-Komplex eines chiralen Derivates[4] der Aminosäure L-Prolin beschichtet sind und die direkte dünnschichtchromatographische Trennung von Enantiomeren nach dem Prinzip der chiralen Ligandenaustauschchromatographie erlauben.[5][6][7] Als weitere stationäre Phasen für die DC eignen sich auch Aluminiumoxid, Magnesiumsilikat, Kieselgur, Polyamid, Cellulose.
Mobile Phase Bearbeiten
Als Laufmittel werden in der Normalphasen-DC unpolare organische Lösungsmittel in der Regel als Gemisch mit mäßig polaren Lösungsmitteln genutzt (z. B. Petrolether und Essigsäureethylester); in der Umkehrphasen-DC dagegen polare Laufmittel (z. B. Acetonitril und Wasser). Über das Mischungsverhältnis kann man die Polarität des Fließmittels steuern.
Die Adsorptionsfähigkeit der funktionellen Gruppen mit Kieselgel nimmt in der Folge -COOH > -OH > -NH2 > SH > -CHO > CO > CO2R > - OCH3 > -HC=CH- ab.
Organische Carbonsäuren oder Alkohole weisen also auf Kieselgel eine sehr hohe Adsorption und damit geringe RF-Werte auf. Bei einem wenig polaren Laufmittel können diese Substanzen möglicherweise am Startfleck verbleiben.
Trennstrecke Bearbeiten
Verschiedene Arten der Diffusion wirken einer guten Auftrennung entgegen. Entscheidend ist der rasche Wechsel der Teilchen zwischen den beiden Phasen. Daher ist es auch günstig, möglichst geringe Laufstrecken und feine, einheitliche Korngrößen des Schichtmaterials zu haben. Zu geringe und zu hohe Geschwindigkeiten der mobilen Phase wirken sich negativ aus. Eine zu geringe Geschwindigkeit begünstigt eine Vergrößerung der Zonen, in denen sich die Probemoleküle aufhalten. Je mehr Zeit zur Verfügung steht, desto größer ist die Rolle, die Diffusionsprozesse innerhalb der mobilen Phase spielen. Ist die Geschwindigkeit zu hoch, kommt es seltener zu einem Wechsel der Teilchen zwischen der mobilen und der stationären Phase. Das führt zu einer größeren statistischen Streuung und ist ebenfalls unerwünscht. Bei allen chromatographischen Methoden gibt es entsprechend eine optimale Geschwindigkeit der mobilen Phase, die durch die Van-Deemter-Gleichung beschrieben wird. Je feiner die Korngrößen sind (bzw. die Dimensionen), desto höher kann die Geschwindigkeit werden. Daraus folgen auch ökonomische Vorteile.
Bei der DC ist die erwünschte räumliche Auftrennung zwischen den verschiedenen Probekomponenten der gesamten Laufstrecke proportional (Abstand Startlinie – Laufmittelfront). Die Vergrößerung der einzelnen Zonen auf Grund statistischer Effekte ist geringer (nicht der Laufstrecke proportional sondern der Wurzel der Laufstrecke). Daher ist es bei schwierigen Trennungen sinnvoll, größere DC-Folien und Laufstrecken zu verwenden.
Mit der Dünnschichtchromatographie lässt sich auf einer 15cm langen Laufstrecke eine Trennleistung von ca. 400–3000 theoretischen Böden erzielen.
Praktische Durchführung Bearbeiten
Probenauftrag Bearbeiten
Die zu untersuchende Substanz wird in einem geeigneten Lösungsmittel gelöst und mit Hilfe einer Kapillare punkt- oder strichförmig aufgetragen. Dies geschieht bei der eindimensionalen DC auf der Startlinie der Folie oder Platte, bei der zweidimensionalen DC (siehe unten) in einer Ecke. Die zu trennende Substanzmenge beträgt ca. 5–20 Mikrogramm. Für eine besonders gleichmäßige und auch quantitativ reproduzierbare Auftragung stehen auch Maschinen zur Verfügung, welche die Lösung mit Hilfe von Druckluft oder Stickstoff aufsprühen. Die stationäre Phase (Trennschicht) besteht aus einer dünnen Schicht eines sehr feinkörnigen Materials (z. B. Kieselgel, Kieselgur, Aluminiumoxid, Cellulose). Diese Trennschicht ist sehr gleichmäßig auf eine Trägerfolie oder Trägerplatte aus Kunststoff, Aluminiumblech oder Glas aufgetragen und kommerziell in unterschiedlichen Schichtdicken erhältlich.
Daneben werden auf der Startlinie in vielen Fällen auch Lösungen von reinen Vergleichssubstanzen oder Vergleichsmischungen aufgetragen. Es ist entscheidend, die Auftragzonen möglichst eng zu halten (wenige Millimeter). Kommerziell erhältlich sind auch DC-Folien mit so genannten Konzentrationszonen. Hier wird eine Zone mit besonders geringer Adsorption vorgeschaltet. Die Probeflecken werden dadurch nach Beginn der Chromatographie in der Richtung der Laufstrecke zusammengestaucht und so sind besonders enge Auftragzonen erzielbar.
Nach dem Auftragen muss die Platte getrocknet werden, da restliches Lösungsmittel das Ergebnis verändern kann.
Für spezielle Anwendungen kann auch das „Waschen“ der Platte vor dem Auftragen der Probe oder auch das Trocknen im Exsikkator oder Trockenschrank bei erhöhter Temperatur notwendig sein. Gewaschen werden die Platten durch Einstellen in eine Chromatographiekammer mit dem entsprechenden Lösungsmittel, bis die Fließmittelfront die Oberkante der Platte erreicht hat.
Auftrennung Bearbeiten

Nach dem Auftragen wird die Platte senkrecht in eine Chromatographiekammer mit einem geeigneten Fließmittel (mobile Phase) eingestellt. Um eine Beeinflussung der Ergebnisse durch das Verdampfen des Fließmittels zu verhindern, führt man die Trennung in einer mit dem Fließmittel gesättigten Atmosphäre in einem geschlossenen Gefäß durch. Zur besseren Sättigung des Dampfraumes mit Laufmittel kann ein Filterpapier eingelegt werden. Die Sättigung verhindert Verdampfen des Laufmittels von der Platte und damit eine Änderung der Zusammensetzung auf der Platte.
Das Fließmittel saugt sich nun über Kapillarkräfte in die stationäre Phase nach oben. Sobald die Flüssigkeit die Startlinie erreicht hat, lösen sich die Substanzen in ihr. Die Moleküle sind nun den Anziehungskräften der stationären Phase einerseits und den Anziehungskräften der mobilen Phase andererseits ausgesetzt. Je nach Kräfteverhältnis bleibt ein Teilchen eher am Startpunkt oder es wandert mit der mobilen Phase nach oben. Im Allgemeinen gilt: Je unpolarer das Fließmittel ist, desto weniger wandern polare Substanzen und umgekehrt. Es gilt aber auch, dass polare Substanzen mit polaren stationären Phasen stärker wechselwirken. Die Lösungsmittelpolarität ist dabei analog wie in der Säulenchromatographie.
Kurz bevor die Laufmittelfront das obere Ende der Platte erreicht, wird die Platte aus der Chromatographiekammer entnommen und möglichst zügig getrocknet.
Auswertung Bearbeiten

Im einfachsten Fall sind die getrennten Substanzen beim Betrachten unter UV-Licht als Punkte sichtbar. Alternativ können sie vor der Chromatographie mit Chromophoren derivatisiert werden, damit sie UV-aktiv werden. Auch das Besprühen mit oder Tauchen in Reagenzienlösungen sind weitere Möglichkeiten.
Viele Schichtmaterialien enthalten Zusätze, die im UV-Licht fluoreszieren und an denjenigen Stellen dunkle Fluoreszenzlöschung zeigen, an denen sich die getrennten Stoffe befinden. Diese Fluoreszenzfarbstoffe dürfen während der chromatographischen Trennung nicht wandern. Gebräuchlich sind vor allem Mangan aktiviertes Zinksilikat (mit UV-Licht der Wellenlänge 254 nm bestrahlt) und Calciumwolframat (mit UV-Licht der Wellenlänge 366 nm bestrahlt). Tatsächlich handelt es sich bei der Methode nicht um eine Fluoreszenzlöschung im engeren Sinne. Probemoleküle werden sichtbar, wenn sie im Bereich von 254 nm oder 366 nm UV-Licht absorbieren. Es gelangt dann weniger UV-Licht zu den Fluoreszenzfarbstoffmolekülen (dunkle Flecken auf grün bzw. blau leuchtendem Hintergrund zu sehen). Dazu müssen genügend viele funktionelle Gruppen bzw. genügend große Systeme mit konjugierten Doppelbindungen vorhanden sein. Gesättigte Kohlenwasserstoffe und viele Aminosäuren sind daher mit dieser Methode nicht nachzuweisen, aromatische Verbindungen z. B. sehr leicht bei 254 nm.
Auch die Eigenfluoreszenz bestimmter Stoffe oder andere Eigenschaften wie Radioaktivität können zur Detektion herangezogen werden.
Bei der Verwendung von Sprüh- oder Tauchreagenzien (z.B. NBD-Cl, Molybdatophosphorsäure oder 2,7-Dichlorfluorescein) laufen Farbreaktionen ab, die empfindlich und spezifisch genug sind, um zum Nachweis bestimmter funktioneller Gruppen verwendet zu werden. Über die Auswahl der Farbreaktion lässt sich der Informationsgehalt bei der DC wesentlich erhöhen. Alternativ werden Reaktionen eingesetzt, die allgemein wirksam sind (zum Beispiel Oxidation mit Hilfe von Salpetersäurelösungen oder Ioddampf). Bei einer Reihe von Farbreaktionen ist es erforderlich, die Folie nach dem Besprühen bzw. der Tauchung zu erhitzen.
Eine weitere, sehr einfache Methode besteht in der Bedampfung mit molekularem Iod. Dazu genügt es, in ein Glasgefäß ein paar Iod-Kristalle zu legen. Sie sublimieren, das heißt sie verdampfen direkt bei Raumtemperatur, bilden einen violetten Dampf von Diiodmolekülen. Durch Einlegen einer DC-Folie in einen solchen Trog werden über Diffusion und Reaktion mit den Molekülen der Substanzflecken binnen kurzer Zeit lockere Komplexverbindungen gebildet (lila oder braun). Vorteil bzw. Nachteil der Methode: die Iod-Verbindungen zerfallen relativ rasch.
In der Biochemie ist eine saure Ninhydrin-Lösung ein häufiges Sprühreagenz, um Aminosäuren zu detektieren. Hierbei wird das Ninhydrin über die Schiffsche Base und durch eine Decarboxylierung sowie Hydrolyse zu Ruhemans Violett. Durch Auftragen von Referenzproben, welche unter gleichen Bedingungen gleich weit wandern wie entsprechende Probekomponenten, kann man das qualitative Auftreten von Stoffen nachweisen. Hierzu wird die Lage der verschiedenen Punkte mit der Lage der Referenzproben verglichen.
Um verschiedene DC vergleichen zu können, werden die so genannten -Werte (Retentionsfaktor, Rückhaltefaktor, ratio of fronts) berechnet. Es handelt sich dabei um das Verhältnis Wanderungsstrecke des Substanzfleckes ( ) zur Wanderungsstrecke des Lösemittels ( ): . Die -Werte sind bei gleichem Plattenmaterial und gleicher Laufmittelzusammensetzung Stoffkonstanten.



Die quantitative Auswertung kann mit einem Densitometer ausgeführt werden. Diese Geräte bieten als sogenannte TLC-Scanner die Möglichkeit der Messung im sichtbaren und im ultravioletten Spektralbereich. Sie können auch zur Messung der Fluoreszenz eingesetzt werden. Auch reguläre Flachbettscanner können für eine densiometrische Auswertung in der DC genutzt werden.[8]
Neue Geräteentwicklungen ermöglichen auch die direkte Kopplung der Dünnschichtchromatographie mit der Massenspektrometrie[9] So kann auch eine zuverlässige Identifizierung der chromatographisch getrennten Komponenten über deren Massenspektrum durchgeführt werden.
Komplexere Techniken und Methoden Bearbeiten
Neben der bisher beschriebenen linearen und eindimensionalen Dünnschichtchromatographie sind für spezielle Anwendungen und Trennaufgaben Techniken entwickelt worden, um beispielsweise komplexere Substanzgemische auftrennen zu können. Die Hochleistungsdünnschichtchromatographie stellt eine solche Weiterentwicklung dar.
Zweidimensionale DC Bearbeiten
Bei der zweidimensionalen DC wird nach der ersten Entwicklung das Laufmittel abgedampft, die Platte um 90° gedreht und − in der Regel in einem anderen Laufmittel − eine zweite Entwicklung durchgeführt. Dadurch kann eine bessere Auftrennung bei Multikomponentengemischen erreicht werden. Die Identifizierung ist aber aufwendiger, da keine Referenzsubstanzen mitlaufen können.
Zirkulare Dünnschichtchromatographie Bearbeiten
Eine alternative Technik zur linearen DC stellt die so genannte zirkulare Dünnschichtchromatographie (abgek. CLC vom engl. Centrifugal Layer Chromatography oder RPC von Rotary Planar Chromatography) dar.[10] Hierbei werden runde Glasscheiben verwendet, die ringförmig mit der stationären Phase beschichtet sind. Die Scheibe wird mit Hilfe eines Elektromotors in rasche und gleichmäßig kontrollierte Rotation versetzt. Die Probelösung wird mit Hilfe einer Pumpe zum inneren Rand der Schicht geleitet, vorher und nachher das entsprechende Fließmittel.
Da die stationäre Phase in der Regel einen Farbstoff enthält, der bei UV-Licht der Wellenlänge 254 nm oder 366 nm fluoresziert, kann der Trennvorgang durch Bestrahlung mit einer UV-Lampe kontrolliert werden. Am Anfang des Trennprozesses befindet sich die Probe in einer wenige Millimeter starken Kreiszone am inneren Rand der Scheibe. Mit fortschreitender Trennung wird das Substanzgemisch der Probe in eine Reihe von Ringen aufgespalten, die getrieben von der Fliehkraft nach außen wandern.
Präparative Dünnschichtchromatographie Bearbeiten
Die DC kann auch präparativ, d.h. zur Reinigung kleiner Substanzmengen genutzt werden. Dann wird sie auch PLC (engl. für Preparative Layer Chromatography) genannt. Hierbei werden in der Regel auf Glasplatten mit dickeren stationären Phasen größere Mengen der zu trennenden Substanzmischung strichförmig aufgetragen. Nach dem Trennlauf befinden sich die voneinander getrennten Substanzen als Linien in verschiedenen Höhen. Die Zielsubstanz kann dann mitsamt des Trägermaterials mechanisch abgeschabt werden. Durch einfache Filtration mit einem geeigneten Lösungsmittel wird sie von der stationären Phase eluiert und so rein erhalten.
Auch von den üblichen (analytischen) DC-Folien mit dünner stationärer Phase lassen sich genügende Mengen Reinsubstanz erhalten, um sie für empfindliche Analyseverfahren wie die Massenspektrometrie oder die Infrarotspektroskopie nutzen zu können.
Die zirkulare DC kann ebenfalls präparativ genutzt werden. Hierbei wird die gewünschte Probenkomponente, nachdem sie am äußeren Rand der Trennschicht angelangt ist, gemeinsam mit dem Laufmittel in einem entsprechende Sammelbehälter aufgefangen.
Um größere Substanzmengen bei weit geringerem apparativen Aufwand zu reinigen nutzt man heute eher säulenchromatographische Techniken wie die Flash-Chromatographie.
Vorteile und Nachteile der Dünnschichtchromatographie Bearbeiten
Im Gegensatz zu den leistungsfähigeren Chromatographie-Verfahren wie Gaschromatographie und Hochleistungsflüssigkeitschromatographie kommt die DC mit geringem apparativen Aufwand aus und stellt sich als schnelles, vielseitiges und preiswertes Analyseverfahren dar.
Die Gaschromatographie lässt sich nur bei Proben anwenden, die unzersetzt verdampfbar sind. Bei der Flüssigchromatographie gibt es wenig Einschränkungen. Fast immer lässt sich ein Weg finden, eine Probe aufzulösen. Im Vergleich zu den säulenchromatographischen Methoden besteht der Vorteil, dass Proben, die Gruppen von Komponenten enthalten, die sich jeweils stark in der Polarität unterscheiden, leichter zu erfassen sind. Laufmittelwechsel ist nicht so leicht möglich wie bei der Säulenchromatographie. Es ist aber möglich, zunächst in einem Laufmittel zu entwickeln und nach Zwischentrocknung in einem anderen (das sich in der Polarität stark unterscheidet).
Nachteilig ist bei der analytischen Anwendung der DC, dass es schwerer ist, eine quantitative Analyse durchzuführen. Bei bestimmten Aufgabenstellungen genügt es allerdings schon, die Mengenverhältnisse abzuschätzen (Fortschritt einer chemischen Reaktion). Die früheren Probleme einer zuverlässigen Quantifizierung konnten in den letzten Jahren durch Entwicklung leistungsfähiger Densitometer – wie oben erwähnt – überwunden werden. Die Qualitätskriterien der quantitativen Auswertung genügen inzwischen den Richtlinien für die Gute Laborpraxis.
Literatur Bearbeiten
- J. Sherma, B. Fried (Hrsg.): Handbook of Thin-Layer Chromatography. In: Chromatographic Science Series. Volume 55. Marcel Dekker, New York, Basel, Hong Kong 1991, ISBN 0-8247-8335-2.
- Elke Hahn-Deinstorp: Applied Thin-Layer Chromatography – Best Practice and Avoidance of Mistakes. Wiley-VCH, Weinheim, New York, Chichester, Brisbane, Singapur, Toronto 2000, ISBN 3-527-29839-8.
Weblinks Bearbeiten
= Säulenchromatographie 0
Die Säulenchromatographie (SC), in der Standardsprache auch Säulenchromatografie (englisch column chromatography), ist ein chromatographisches Trennverfahren, das auf dem Adsorptionsverfahren beruht.
Die Säulenchromatographie eignet sich sehr gut zur Auftrennung eines organischen Stoffgemisches in seine Einzelkomponenten im Gramm-Maßstab.
Durchführung Bearbeiten
Normalerweise wird die Säulenchromatographie derart ausgeführt, dass man zunächst einen Feststoff (beispielsweise Kieselgel oder Aluminiumoxid) mit einem Lösungsmittel aufschlämmt und dann in ein senkrecht stehendes Glasrohr füllt. Das untere Ende des Glasrohres ist konisch verengt und am unteren Ende befindet sich ein regelbares Drehventil zum Ablass der Flüssigkeit. Im konisch verengten Teil wird ein Wattebausch oder eine Glasfritte eingesetzt, damit der Feststoff nicht durch das Rohr ablaufen kann. Das Säulenmaterial muss so beschaffen sein, dass es die zu trennenden Substanzen – je nach Polarität der funktionellen Gruppen – unterschiedlich lange festhält.
Das Lösungsmittel lässt man bis zum oberen Rand der Feststofffüllung ablaufen und gibt dann die zu trennende Substanz in Reinform oder wenig verdünnt mit Lösungsmittel auf den oberen Teil der Feststoffschicht der Säule auf, lässt dann die Substanz in die Feststoffschicht durch Betätigen des Ablassventils leicht einsickern, füllt dann mit Lösungsmittel auf. Durch Wechselwirkung mit dem festen Adsorptionsmaterial wandern die Komponenten als Zonen durch die Säule. Mit einem Lösungsmittel, das beispielsweise über einen Tropftrichter stetig nachläuft, können die Komponenten am unteren Auslass aufgefangen werden.
Im Verlauf der Chromatographie wird sukzessiv meist auch das Lösungsmittel durch andere zunehmend polarere Lösungsmittel ersetzt, damit die Verbindungen, die stärker vom Säulenmaterial festgehalten werden, von der Säule herunterlaufen und nachgewiesen werden können. In mehreren Bechergläsern oder Reagenzgläsern können die einzelnen Fraktionen dann auf Reinheit und chemische Zusammensetzung geprüft werden. Als stationäre Phase eignen sich beispielsweise Kieselgel oder Aluminiumoxid.
Die Lösungsmittelpolarität ändert sich in der Reihenfolge:
Petrolether (n-Pentan/Hexan) < Benzol < Dichlormethan < Trichlormethan < Diethylether < Essigsäureethylester < Aceton < Ethanol
Um stark polare Verbindungen zu trennen oder von der beschickten Säule zu eluieren, muss die Polarität des Lösungsmittels gesteigert werden oder eine geeignete Mischung aus mehreren Lösungsmitteln verwendet werden.
Die Vorproben mittels der Dünnschichtchromatographie mit entsprechenden Lösungsmittelmischungen können meist direkt auf die säulenchromatographische Trennung verwendet werden. Auch kann die Dünnschichtchromatographie zum Test der abgelaufenen Fraktionen auf Reinheit verwendet werden.
Schneller sind derartige Trennungen durch Einsatz von Druckluft durchführbar (Flash-Chromatographie). Die Druckluft drückt auf die überstehende Lösungsmittelschicht über dem Kieselgel, beschleunigt den Durchlauf und verkürzt somit beträchtlich die Trennzeit.
Literatur Bearbeiten
- Ullmanns Enzyklopädie der technischen Chemie. 3. Auflage, Band 2, S. 106–112.
Schutzgruppe Bearbeiten


Eine Schutzgruppe (engl. protecting group – daher häufig als allgemeine Abkürzung in Formelschemata PG) ist in der Chemie ein Substituent, der während einer komplizierteren, mehrstufigen chemischen Synthese in ein Molekül eingeführt wird, um eine bestimmte funktionelle Gruppe vorübergehend zu schützen und so eine unerwünschte Reaktion zu verhindern. Nach der Durchführung der gewünschten Reaktion an anderer Stelle des Moleküls wird die Schutzgruppe wieder abgespalten. Für viele funktionelle Gruppen sind mehrere mögliche Schutzgruppen bekannt, die sich in ihrer Stabilität und den Bedingungen für ihre Abspaltung unterscheiden.
Bei der Synthese von speziellen Verbindungsklassen mit sich wiederholenden funktionellen Gruppen – in der Regel sind dies Biomoleküle wie Peptide, Oligosaccharide oder Nukleotide – haben sich Standardsätze an Schutzgruppen etabliert. Schutzgruppen sind heute ein wichtiges Werkzeug in der Synthese von komplexen Verbindungen geworden.
Die Anforderungen an eine Schutzgruppe sind recht hoch. Dazu gehört, dass sie sich mit sehr guter Ausbeute und spezifisch an eine funktionelle Gruppe einführen lassen und ebenso unter milden Bedingungen wieder abzuspalten sein muss. Für beide Schritte sollten die Reaktionsbedingungen standardisierbar sein. Zudem muss die Schutzgruppe unter möglichst vielen Reaktionsbedingungen stabil sein. Nach Möglichkeit sollten die entstehenden Reaktionsprodukte leicht abtrennbar sein, und optimalerweise ist das Schutzgruppen-Reagenz auch noch preiswert. Je breiter der Erfahrungsschatz mit einer Schutzgruppe ist, umso besser ist die Vorhersagbarkeit der Reaktivität der Schutzgruppe.
Geschichte Bearbeiten


Die Geschichte der Schutzgruppentechnik ist untrennbar verbunden mit der gezielten Verwendung verschiedener Ausgangsverbindungen für die Synthese eines Zielmoleküls. Die frühen Schutzgruppen beruhten in der Regel darauf, dass die Ausgangsverbindung so gewählt wurde, dass eine reaktive funktionelle Gruppe durch einen Rest blockiert und somit unreaktiv war. So wurden z. B. Anisole anstatt Phenole gewählt oder Ester anstelle von freien Alkoholgruppen. Erst mit der ab Anfang des 20. Jahrhunderts aufkommenden gezielten Synthese von immer komplexer werdenden Verbindungen wurde die Schutzgruppentechnik wirklich bedeutsam. Etwa ab 1960 wurde begonnen, in die Chemie der Schutzgruppen erheblichen Forschungsaufwand zu investieren. Während dieser Zeit begannen Chemiker immer komplexere Naturstoffe zu synthetisieren. Hervorzuheben sind vor allem die damaligen Arbeiten der Nobelpreisträger Robert B. Woodward, Elias J. Corey und Albert Eschenmoser, die bei der Synthese von komplexen Naturstoffen Pionierarbeit geleistet haben.[11][12]
Heute gibt es eine Vielzahl von Schutzgruppen, die in Monographien bezüglich ihrer Eigenschaften zusammengefasst sind.[13][14] Dabei gibt es neben etablierten Schutzgruppen sehr viele exotische Schutzgruppen, die nur für eine Synthese oder ein recht spezielles Gebiet entwickelt wurden.
Anforderungen an eine Schutzgruppe Bearbeiten
Das Einfügen und Entfernen von Schutzgruppen stellen keine produktiven Reaktionen in einer Abfolge von Syntheseschritten dar, ihr Produkt kommt dem angestrebtem Endprodukt der Synthese nicht näher. Deswegen werden an Schutzgruppenreaktionen oft hohe Anforderungen bezüglich Preis, Ausbeute und Entwicklungsaufwand für die Reaktion gestellt.
Als Grundanforderungen für eine gute Schutzgruppe haben sich folgende Merkmale herausgebildet:
- Das Reagenz muss käuflich und preiswert oder leicht herstellbar sein
- Die Schutzgruppe muss einfach, spezifisch und in hohen Ausbeuten einführbar sein
- Sie muss stabil gegenüber einer möglichst großen Anzahl an Reaktionsbedingungen und Aufarbeitungs- und Reinigungsmethoden sein
- Sie muss spezifisch, hoch selektiv und in hohen Ausbeuten abspaltbar sein. Dabei sollten die Bedingungen standardisierbar sein.
- Sie darf kein neues Stereozentrum und auch kein diastereotopes Zentrum bilden
- Sie sollte einfach in NMR-Spektren erkennbar sein und möglichst wenig durch Signalüberlappung stören
Einen sehr wichtigen Aspekt stellt die hohe Selektivität der Abspaltung dar denn es müssen häufig unterschiedliche funktionelle Gruppen unabhängig von einander geschützt und entschützt werden. Im Idealfall ist immer nur eine von vielen Schutzgruppen vom Abspaltungsprozess betroffen. Das Verhalten von Schutzgruppen in der Praxis lässt sich, besonders wenn mehrere verschiedene Schutzgruppen in einem Molekül verwendet werden, nicht immer anhand der Literatur korrekt vorhersagen. Daher muss in manchen Fällen trotz großem Erfahrungsschatz sowohl für das Einführen als auch für das Abspalten noch erhebliche Entwicklungsarbeit während einer Synthese geleistet werden.[15][16]
Orthogonalität von Schutzgruppen Bearbeiten

Orthogonalität von Schutzgruppen bedeutet, dass sich bei Verwendung mehrerer Schutzgruppen verschiedenen Typs jede Schutzgruppe einzeln und in einer beliebigen Reihenfolge aufgrund der verschiedenen Abspaltreagenzien abspalten lässt, ohne dass eine der anderen Schutzgruppen angegriffen wird. Im gezeigten Beispiel der geschützten Aminosäure Tyrosin kann der Benzylester hydrogenolytisch, die Fluorenylmethylenoxy-Gruppe (Fmoc) durch Basen (wie z. B. Piperidin) und der phenolische tert.-Butylether mit Säuren (z. B. mit Trifluoressigsäure) gespalten werden.
Ein weit verbreitetes Beispiel für diese Anwendung ist die Fmoc-Peptidsynthese, die sowohl in Lösung als auch auf fester Phase eine große Bedeutung erlangt hat.[17] Die Schutzgruppen in der Festphasensynthese müssen bezüglich der Reaktionsbedingungen wie Reaktionszeit, Temperatur und Reagenzien so standardisiert sein, dass sie von einem Automaten durchgeführt werden und dabei Ausbeuten von weit über 99 % erreicht werden können, da sonst die Trennung des resultierenden Gemisches der Reaktionsprodukte praktisch unmöglich ist.[18]
-
 Schematische Darstellung einer Festphasen-Peptidsynthese mit orthogonalen Schutzgruppen X und Y
Schematische Darstellung einer Festphasen-Peptidsynthese mit orthogonalen Schutzgruppen X und Y -
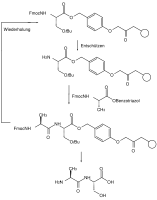 Fmoc-Festphasen Peptidsynthese mit orthogonalen Schutzgruppen
Fmoc-Festphasen Peptidsynthese mit orthogonalen Schutzgruppen



Eine weitere wichtige Anwendung von orthogonalen Schutzgruppen ist die Kohlenhydrat-Chemie. Da Kohlenhydrate über Hydroxygruppen mit sehr ähnlicher Reaktivität verfügen, muss für eine gezielte synthetische Umsetzung der Schutz bzw. das Entschützen von einzelnen Hydroxygruppen möglich sein. Einen ähnlichen Fall stellt die Synthese von Nukleotiden dar. Hier hat man zum einen das Problem (wie bei der Peptidsynthese), dass es sich um vektorielle Moleküle handelt. Zum anderen hat man hier auch das Problem der Kohlenhydratchemie mit dem Zuckerrest der Ribose bei der Synthese von RNA-Molekülen.
Aber auch in der Synthese von komplexen Naturstoffen oder Wirkstoffen mit vielen funktionellen Gruppen ist man auf die Orthogonalität der Schutzgruppen angewiesen.[12][19]
Labilität bzw. Abspaltung von Schutzgruppen Bearbeiten
Bei Schutzgruppen haben sich verschiedene Reaktionsbedingungen etabliert, die dem Orthogonalitäts-Prinzip entsprechen, unter denen Schutzgruppen abgespalten werden. Man kann grob zwischen folgenden Abspaltbedingungen unterscheiden:[20]
- Säurelabile Schutzgruppen
- Basenlabile Schutzgruppen
- Fluoridlabile Schutzgruppen
- Enzymlabile Schutzgruppen
- Reduktionslabile Schutzgruppen
- Oxidationslabile Schutzgruppen
- Schutzgruppen die durch Schwermetallsalze oder deren Komplexe gespalten werden
- Photolabile Schutzgruppen
- Zwei-Stufen-Schutzgruppen
Säurelabile Schutzgruppen lassen sich durch die Einwirkung von Säuren abspalten. Die Triebkraft ist hier häufig die Bildung eines verhältnismäßig stabilen Carbokations oder ein säurekatalysiertes Gleichgewicht, das auf der Seite der freien funktionellen Gruppe liegt. Beispiele für säurelabile Schutzgruppen sind die tert.-Butylester, -ether und -carbamate, welche stabile Kationen bilden, und die Acetale, bei denen in Anwesenheit von Wasser das säurekatalysierte Gleichgewicht auf der Seite der entsprechenden Aldehyde oder Ketone liegt.


Bei den basenlabilen Schutzgruppen kann man mechanistisch zwischen der basischen Hydrolyse und der baseninduzierten β-Eliminierung unterscheiden. Carbonsäureester (mit Ausnahme der tert.-Butylester) werden von Hydroxidionen nukleophil angegriffen und so hydrolytisch gespalten. Amide hingegen werden selten so gespalten, da sie recht harsche Bedingungen benötigen. Eine Ausnahme stellt hier die Phthaloyl-Gruppe dar, da diese mit Hydrazin schon unter recht milden Bedingungen gespalten wird. Bei der β-Eliminierung läuft eine Reaktionskaskade ab: Zunächst wird ein Proton durch die Base abgespalten und ein Carbanion gebildet. Durch eine geeignete Abgangsgruppe wird nun die Schutzgruppe unter Bildung einer Vinylverbindung gespalten. Zum letzteren Fall zählt vor allem die Fmoc-Gruppe.

Fluorid-Ionen bilden mit Silicium eine sehr stabile Bindung. Daher werden die Silicium-Schutzgruppen praktisch ausnahmslos durch Fluorid-Ionen gespalten. Je nach Art des Gegenions bzw. des Abspaltreagenzes können jedoch auch verschiedene Silicium-Schutzgruppen in Abhängigkeit von der sterischen Hinderung des Silicium-Atoms selektiv gespalten werden. Der Vorteil von Fluorid-labilen Schutzgruppen ist, dass keine andere Schutzgruppe unter den Abspaltbedingungen angegriffen wird.
Ester können häufig durch Enzyme wie Lipasen gespalten werden. Da Enzyme bei einem pH-Wert zwischen 5 und 9 und bei moderaten Temperaturen von etwa 30–40 °C arbeiten und zudem, was die Carbonsäure betrifft, noch sehr selektiv sind, ist diese Methode eine zwar selten benutzte, aber sehr attraktive Methode zur Schutzgruppenspaltung.
Benzylgruppen können reduktiv durch katalytische Hydrierung gespalten werden. Zum Einsatz kommen Benzylgruppen als Ether, Ester, Urethane, Carbonate oder Acetale und werden zum Schutz von Alkoholen, Carbonsäuren, Aminen und Diolen eingesetzt.

Nur wenige Schutzgruppen, die oxidativ entfernt werden können, sind gebräuchlich. Es handelt sich hier in der Regel um Methoxybenzylether. Sie können mit Cerammmoniumnitrat (CAN) oder Dichlordicyanobenzochinon (DDQ) über ein Chinomethin gespalten werden.
Die Doppelbindung eines Allylrestes kann durch Platingruppenelemente (wie Palladium, Iridium oder Platin) zur Vinylverbindung isomerisiert werden. Die so erhaltenen Enolether bei geschützten Alkoholen oder Enamine bei geschützten Aminen können leicht sauer hydrolysiert werden.
Photolabile Schutzgruppen enthalten ein Chromophor, das durch Bestrahlung mit einer geeigneten Wellenlänge aktiviert und so abgespalten werden kann.[21] Als Beispiel sei hier die o-Nitrobenzylgruppe (ONB) aufgeführt.

Eine besondere Form von Schutzgruppen stellen die Zwei-Stufen-Schutzgruppen dar. Diese zeichnen sich durch eine hohe Stabilität aus, da die Schutzgruppe zunächst durch eine chemische Transformation in eine abspaltbare Gruppe umgewandelt werden muss. Diese Art an Schutzgruppen finden jedoch selten Anwendung, da hier ein zusätzlicher Aktivierungsschritt notwendig ist, was die Synthese um eine weitere Stufe verlängert.
Funktionelle Gruppen Bearbeiten
Amine Bearbeiten
Für die Aminofunktion ist die bei weitem größte Vielfalt an Schutzgruppen verfügbar. Dies hängt zum einen damit zusammen, dass Aminen in der Peptidsynthese eine besondere Wichtigkeit zukommt, aber auch an ihren Eigenschaften: Sie sind zum einen recht potente Nukleophile, aber auch verhältnismäßig starke Basen. Diese Eigenschaften führten dazu, dass immer neue Schutzgruppen für Amine entwickelt wurden.[22]
Viele Schutzgruppen für Amine basieren auf Carbamaten. Diese lassen sich leicht in Form von Carbonsäurechloriden einführen. Ihre Triebkraft bei der Spaltung beziehen sie durch die Bildung des sehr stabilen Kohlenstoffdioxid-Moleküls. Basierend auf unterschiedlichen Resten am Carbamat wurden verschiedene Spaltungsmöglichkeiten entwickelt. Die am häufigsten benutzten Carbamate sind die tert.-Butyloxycarbonyl–, Benzyloxycarbonyl–, die Fluorenylmethylenoxycarbonyl– und die Allyloxycarbonyl–Verbindungen.
| Rest | Formel | Name | Abkürzung | Spaltung |
|---|---|---|---|---|
| tert.-Butyl |  |
tert.-Butyloxycarbonyl | Boc | sauer; Trifluoressigsäure (TFA) rein oder als Lösung in Dichlormethan[23], 3 M Salzsäure in Essigsäureethylester[24] oder 10 %ige Schwefelsäure in Dioxan[25] |
| Benzyl |  |
Benzyloxycarbonyl | Cbz oder Z | hydrogenolytisch; Wasserstoff und Palladium auf Aktivkohle[26], Lithium oder Natrium in flüssigem Ammoniak[27] |
| Fluorenylmethylen |  |
Fluorenylmethylenoxycarbonyl | Fmoc | basisch; 20–50 % Piperidin in Dimethylformamid[28] |
| Allyl |  |
Allyloxycarbonyl | Alloc | übergangsmetallkatalysierte Spaltung ; Metalle wie Palladium(0)– oder Nickel(0)-Komplexe[29] |
Neben den Carbamaten sind noch eine Reihe anderer N-Acyl-Derivate als Schutzgruppen von Bedeutung, aber bei weitem nicht so weit verbreitet. Dazu gehören beispielsweise die Phthalimide, die entweder durch die Umsetzung der primären Amine mit Phthalsäureanhydrid oder durch den Aufbau der Aminogruppe über eine Gabriel-Synthese zugänglich sind. Die Spaltung der Phthalimide erfolgt normalerweise durch Hydrazinhydrat oder Natriumboranat.[30] Trifluoracetamide sind überaus leicht im basischen zu verseifen, daher dienen die durch die Umsetzung mit Trifluoressigsäureanhydrid erhaltenen Acetamide gelegentlich als Schutzgruppe für Amine.
Bei Indolen, Pyrrol und Imidazolen, also heterocyclischen Verbindungen, finden die bei normalen Aminen für eine Schutzgruppe häufig zu stabilen N-Sulfonyl-Derivate ihre Anwendung. Die Darstellung erfolgt hier durch Sulfonierung mit Phenylsulfonylchlorid und dem deprotonierten Heterocyclus. Die Spaltung erfolgt durch basische Hydrolyse. N-Acyl-Derivate von primären und sekundären Aminen sind zwar relativ leicht durch die Umsetzung der Amine mit einem Arylsulfonsäurechlorid zugänglich, können aber nur schwer z. B. unter den Bedingungen einer Birch-Reduktion (Natrium in flüssigem Ammoniak) oder durch Umsetzung mit Natriumnaphthalid gespalten werden.[31]
Unter den N-Alkyl-Derivaten haben die durch Alkylierung oder reduktive Alkylierung darstellbaren N-Benzyl-Derivate eine gewisse Bedeutung. Die Spaltung erfolgt wie bei der Cbz-Gruppe reduktiv und normalerweise durch katalytische Hydrierung oder durch Birch-Reduktion. N-Alkyl-Amine haben hier den entscheidenden Nachteil gegenüber den Carbamaten oder Amiden, dass der basische Stickstoff erhalten bleibt.
Alkohole Bearbeiten
Die klassische Schutzgruppe für Alkohole sind Carbonsäureester. Häufig sind die Ester von Vorstufen käuflich erhältlich oder können leicht durch Umsetzung der Alkohole mit den Säurechloriden oder Anhydriden durch eine Schotten-Baumann-Reaktion oder aber durch Umesterung erhalten werden. Die Spaltung der Ester erfolgt in der Regel durch die Umsetzung mit Nukleophilen wie den Alkalihydroxiden, Alkali–Alkoholaten oder Lithium– bzw. Magnesium–organischen Verbindungen; alternativ auch reduktiv durch Umsetzung mit komplexen Hydriden wie Lithiumaluminiumhydrid. Die Reaktivität der Ester gegenüber nukleophilen Angriffen sinkt mit der sterischen Hinderung der Carbonsäure in der Reihenfolge:

Die Reaktivität der Alkohole sinkt ebenfalls mit der steigenden sterischen Hinderung der Alkohole:



Die wichtigsten Ester, die als Schutzgruppen gebräuchlich sind, sind die Essigsäureester, die Benzoesäureester und die Pivalinsäureester, da diese sich nach den angegebenen Reaktivitäten differenziert voneinander abspalten lassen.
Zu den wichtigsten Schutzgruppen von Alkoholen und auch Phenolen zählen die sehr gut untersuchten und dokumentierten trisubstituierten Silylether. Dabei trägt das Silicium als organische Reste sowohl Alkyl- als auch Arylgruppen. Dieser Typ an Schutzgruppe hat den Vorteil, dass er bezüglich der Einführung und besonders bezüglich der Abspaltung sehr gut moderierbar ist. Hergestellt werden diese Ether entweder in einer Williamson-Ethersynthese aus dem Chlorsilan und einem Alkoholat-Ion oder aber durch die Verwendung von Aktivierungsreagenzien wie Imidazol.
Für rein analytische Zwecke, z. B. um ein Kohlenhydrat flüchtig zu machen und mit Hilfe von GC-MS detektieren zu können, existieren kommerziell erhältliche Reaktionskits.[32] Silylether sind grundsätzlich empfindlich gegenüber Säuren und Fluorid-Ionen. Letzteres wird meist für deren Spaltung ausgenutzt. Die kommerziellen Preise der Chlorsilane sind jedoch je nach Substitution sehr unterschiedlich. Das preiswerteste Chlorsilan ist hier das Chlortrimethylsilan (TMS-Cl), das ein Abfallprodukt der Silikon-Herstellung nach Rochow und Müller ist. Eine andere gebräuchliche Quelle der Trimethylsilyl-Gruppe ist das Hexamethyldisilazan (HMDS). Jedoch sind die Trimethylsilylether auch extrem empfindlich gegenüber sauerer Hydrolyse (beispielsweise ist schon Kieselgel als Protonendonator ausreichend) und werden daher heute selten als Schutzgruppe benutzt.
| Name | Formel | Abkürzung | Spaltung |
|---|---|---|---|
| Trimethylsilyl |  |
TMS | Kaliumfluorid, Essigsäure oder Kaliumcarbonat in Methanol[33] |
| Triethylsilyl |  |
TES | 10–100 mal stabiler als eine TMS-Gruppe;[34] Trifluoressigsäure in Wasser/Tetrahydrofuran,[35] Essigsäure in Wasser/Tetrahydrofuran,[36] Fluorwasserstoffsäure, Pyridiniumhydrofluorid in Pyridin[37] |
| tert.-Butyldimethylsilyl |  |
TBS, TBDMS | Essigsäure in Tetrahydrofuran/Wasser,[38] Pyridiniumtosylat in Methanol,[39] Trifluoressigsäure in Wasser,[40] Fluorwasserstoffsäure in Acetonitril,[41] Pyridiniumhydrofluorid in Tetrahydrofuran,[42] Tetrabutylammoniumfluorid in THF[43] |
| Triisopropylsilyl |  |
TIPS | Unter gleichen Bedingungen wie TBS aber längere Reaktionszeiten; Tetrabutylammoniumfluorid in Tetrahydrofuran, Fluorwasserstoffsäure in Acetonitril, Pyridiniumhydrofluorid in Tetrahydrofuran.[44] |
| tert.-Butyldiphenylsilyl |  |
TBDPS | Unter gleichen Bedingungen wie TBS aber längere Reaktionszeiten (100–250 mal langsamer als TBS und 5–10 mal langsamer als TIPS); Tetrabutylammoniumfluorid in Tetrahydrofuran, Fluorwasserstoffsäure in Acetonitril, Pyridiniumhydrofluorid in Tetrahydrofuran[45] |
Eine weitere Klasse an Schutzgruppen für Alkohole sind die Alkylether. Auch hier gibt es vielfältige und orthogonale Möglichkeiten die Ether zu spalten. Aliphatische Methoxyether sind nur schwer und unter drastischen Bedingungen spaltbar, so dass diese im Allgemeinen nur bei Phenolen zum Einsatz kommen.
| Name | Formel | Abkürzung | Spaltung |
|---|---|---|---|
| Methyl | Me | In der Regel nur für Phenole gebräuchlich; Iodtrimethylsilan in Chloroform, Dichlormethan oder Acetonitril,[46] Bortribomid oder Bortrichlorid in Dichlormethan,[47] Lewis-Säuren (Aluminiumchlorid, Bortrifluorid in Gegenwart von Thiolen)[46] | |
| Benzyl |  |
Bn | reduktiv; Katalytische Hydrierung (Palladium auf Aktivkohle, Raney-Nickel oder Rhodium auf Aluminiumoxid als Katalysator)[48] |
| p-Methoxybenzyl | PMB, MPM | oxidativ; DDQ (Dichlordicyanochinon) in Dichlormethan,[49] Cerammoniumchlorid in Wasser[50] | |
| 3,4-Dimethoxbenzyl |  |
DMB, DMPM | wie PMB oxidativ; DDQ (Dichlordicyanochinon) in Dichlormethan, Cerammoniumchlorid in Wasser[51] |
| Triphenylmethyl (Trityl) |  |
Tr | sauer; Ameisensäure in Ether oder Wasser,[52] 80 % Essigsäure,[53] 1 M Salzsäure[54] |
| tert.-Butyl |  |
sauer; wasserfreie Trifluoressigsäure, Bromwasserstoffsäure/Essigsäure, 4 N Salzsäure[55] | |
| Allyl | Kalium-tert.-butanolat,[56] Palladium auf Aktivkohle, DABCO in Methanol, diverse Platin-Element-Komplexe – anschließend saure Aufarbeitung.[57] | ||
| Allyloxycarbonyl | Alloc | Wie Allyl; Kalium-tert.-butanolat, Palladium auf Aktivkohle, DABCO in Methanol, diverse Platin-Element-Komplexe – anschließend saure Aufarbeitung[58] | |
| Methoxymethyl | MOM | Sauer; 6 M Salzsäure in Tetrahydrofuran/Wasser[59] | |
| Methylthiomethyl | MTM | Quecksilber(II)-chlorid/Calciumcarbonat in Acetonitril/Wasser,[60] Silbernitrat in Tetrahydrofuran/Wasser[61] | |
| (2-Methoxyethoxy)methyl | MEM | Wässrige Bromwasserstoffsäure in Tetrahydrofuran,[62] Zinkbromid in Dichlormethan[63] | |
| Benzyloxymethyl | BOM | Vergleichbar mit der Stabilität von MOM, MEM und SEM;[64] Reduktiv; Natrium in flüssigem Ammoniak,[65][66] Katalytische Hydrierung (Palladiumhydroxid auf Aktivkohle), Raney-Nickel in Ethanol[67][68] | |
| β-(Trimethylsilyl)ethoxymethyl |  |
SEM | Labiler als MEM und MOM gegenüber saurer Hydrolyse; 0.1 M Salzsäure in Methanol,[69] konzentrierte Fluorwasserstoffsäure in Acetonitril,[39] Bortrifluorid-Etherat in Dichlormethan,[70] Tetrabutylammoniumfluorid in HMPT (Hexamethylphosphorsäuretriamid) oder in Tetrahydrofuran[71][72] |
| Tetrahydropyranyl |  |
THP | Essigsäure in Tetrahydrofuran/Wasser,[73] p-Toluolsulfonsäure in Methanol[74] |
1,2-Diole Bearbeiten
Eine besondere Klasse von Alkoholen in der Schutzgruppen-Chemie stellen die 1,2-Diole (Glykole) dar. Die Nachbarstellung von zwei Hydroxygruppen kann man z. B. bei Zuckern dazu ausnutzen, dass man beide Hydroxylgruppen abhängig voneinander als Acetal schützt. Gebräuchlich sind hier die Benzyliden–, Isopropyliden– und Cyclohexyliden– bzw. Cyclopentyliden-Acetale.

Die Herstellung der Acetale erfolgt in der Regel durch Verschieben des Gleichgewichtes eines Gemisches des Glycols mit der Carbonyl-Komponente durch Entfernen des Reaktionswassers oder durch Umacetalisierung mit einem einfachen Acetal und dem Entfernen des entstehenden Alkohols aus dem Reaktionsgemisch.

Gerade in der Zuckerchemie wird hier die unterschiedliche Stellung der Hydroxylgruppen zueinander ausgenutzt, um diese in bestimmter stereochemischer Abhängigkeit selektiv zu schützen. So reagieren (neben den anderen möglichen Kombinationen) die beiden benachbarten Hydroxygruppen bevorzugt miteinander, welche die stabilste Konformation bilden.[75][76]

Acetale können grundsätzlich in wässrigen sauren Lösungsmitteln wieder gespalten werden. Einen besonderen Fall stellt hier die Benzyliden-Schutzgruppe dar, die auch reduktiv gespalten werden kann. Dies erfolgt entweder durch katalytische Hydrierung oder durch den Hydriddonor Diisobutylaluminiumhydrid (DIBAL). Die Spaltung durch DIBAL entschützt jedoch nur eine Alkoholgruppe, da der Benzylrest auf der zweiten und sterisch gehinderteren Hydroxylgruppe als Benzylether verbleibt.[77][78]

Carbonylgruppen Bearbeiten
Carbonylgruppen sind vor allem durch nukleophile Angriffe wie Grignard-Reagenzien oder von Hydrid-Ionen gefährdet. Aldehyde können zusätzlich noch zu Carbonsäuren oxidiert werden. Aber auch unerwünschte Reaktionen, die durch säure- und basen-katalysierte Reaktionen der Carbonylgruppe wie z. B. Aldolreaktionen können durch eine geeignete Schutzgruppe verhindert werden.
Die gebräuchlichsten Schutzgruppen für Carbonylgruppen sind Acetale und hier besonders cyclische Acetale mit Diolen. Daneben werden auch cyclische Acetale mit 1,2-Hydroxythiolen oder Dithioglycolen verwendet – die sogenannten O,S– bzw. S,S-Acetale.


Für Acetale als Schutzgruppe für Carbonylverbindungen gilt grundsätzlich das gleiche wie für Acetale als Schutzgruppe für 1,2-Diole. Sowohl die Herstellung als auch die Spaltung sind naturgemäß identisch. Allerdings spielt bei Acetalen als Schutzgruppe der Prozess einer Umacetalisierung eine untergeordnete Rolle, und sie werden in der Regel aus den Glycolen durch Wasserabspaltung hergestellt. Modernere Varianten verwenden hier auch Glycole, bei welchen die Hydroxyl-Wasserstoff-Atome durch eine Trimethylsilyl-Gruppe ersetzt wurden.[79][80] Normalerweise finden einfache Glycole wie das Ethylenglycol oder das 1,3-Propandiol als Diole für die Acetale Verwendung.
Acetale können unter sauren wässrigen Bedingungen gespalten werden. Dabei werden als Säuren die Mineralsäuren verwendet. Cosolvenz ist häufig Aceton, das als Lösungsvermittler benutzt wird. Als nicht-saure Abspaltmethode kann mit einem Palladium(II)-chlorid-Acetonitril-Komplex in Aceton[81] oder mit Eisen(III)-chlorid auf Kieselgel, aufgezogen in Chloroform,[82] gearbeitet werden.
Cyclische Acetale sind sehr viel stabiler gegenüber saurer Hydrolyse als acyclische Acetale. Daher werden praktisch ausschließlich acyclische Acetale benutzt, wenn eine sehr milde Anspaltung nötig ist oder wenn zwei verschiedene geschützte Carbonylgruppen bezüglich ihrer Freisetzung differenziert werden müssen.[83]
Acetale finden jedoch neben ihrer alleinigen Funktion als Schutzgruppe zusätzlich noch Anwendung als chirales Hilfsreagenz. So können Acetale von chiralen Glycolen wie z. B. Derivate der Weinsäure mit hoher Selektivität asymmetrisch geöffnet werden. Dies ermöglicht den Aufbau neuer Chiralitätszentren.[84]

Neben den O,O-Acetalen haben auch noch die S,O- und S,S-Acetale eine, wenn auch geringere, Bedeutung als Carbonylschutzgruppe. Thiole, die man zur Herstellung dieser Acetale einsetzen muss, haben einen sehr unangenehmen Geruch und sind giftig, was die Anwendung sehr einschränkt. Thioacetale und die gemischten S,O-Acetale sind, verglichen mit den reinen O,O-Acetalen, sehr viel stabiler gegenüber saurer Hydrolyse. Dies ermöglicht die selektive Spaltung dieser in Gegenwart von Schwefel-geschützten Carbonylgruppen.
Die Herstellung der S,S-Acetale erfolgt normalerweise analog der O,O-Acetale durch saure Katalyse aus den Dithiolen und der Carbonylkomponente. Aufgrund der großen Stabilität der Thioacetale liegt das Gleichgewicht auf der Seite der Acetale. Es muss zum Unterschied zu den O,O-Acetalen kein Reaktionswasser entfernt werden, um das Gleichgewicht zu verschieben.[85]
S,O-Acetale werden um den Faktor 10.000 schneller hydrolysiert als die entsprechenden S,S-Acetale. Ihre Herstellung erfolgt in Analogie zu diesen aus den Thioalkoholen. Auch ihre Spaltung erfolgt unter vergleichbaren Bedingungen und vorwiegend durch Quecksilber(II)-Verbindungen in wässrigem Acetonitril.[86]
Für Aldehyde ist eine temporäre Schützung der Carbonylgruppe in Anwesenheit von Ketonen als Halbaminal-Anion beschrieben. Hier wird ausgenutzt, dass Aldehyde eine sehr viel höhere Carbonylaktivität aufweisen als Ketone und dass viele Additionsreaktionen reversibel sind.[87][88]

Carboxygruppen Bearbeiten
Die wichtigsten Schutzgruppen für Carboxy-Gruppen sind die Ester von verschiedenen Alkoholen. Daneben sind auch noch Ortho-Ester und Oxazoline in Gebrauch, aber von untergeordneter Bedeutung. Für die Herstellung von Carbonsäureestern gibt es grundsätzlich verschiedene Methoden:[89]
- Direkte Veresterung von Carbonsäuren und Alkoholkomponente. Aufgrund der ungünstigen Gleichgewichtslage bei der Reaktion zwischen Alkoholen und Carbonsäuren muss das Gleichgewicht entweder durch das Entfernen des Reaktionswassers oder aber durch das Arbeiten mit großen Überschüssen an Alkohol erfolgen. Dazu muss der Alkohol jedoch sehr preiswert sein. Diese Reaktion ist säurekatalysiert (Schwefelsäure, p-Toluolsulfonsäure oder saure Ionentauscher sind die gebräuchlichsten Veresterungskatalysatoren).
- Die Reaktion von Säureanhydriden oder Säurechloriden mit Alkoholen in Gegenwart von Hilfsbasen. Als Hilfsbase findet hier häufig Pyridin, Diisopropylethylamin oder Triethylamin Anwendung. Diese Reaktion kann mit 4-N,N-Dimethylaminopyridin katalysiert werden, was die Reaktionsgeschwindigkeit im Vergleich zu reinem Pyridin um den Faktor 104 steigert. Im Vergleich zur direkten Veresterung erfolgen diese Methoden unter recht milden Bedingungen.
- Die Reaktion von Carbonsäuresalzen mit Alkylhalogeniden ist eine weitere Methode zur Herstellung von Carbonsäureestern.
- Die Reaktion von Carbonsäuren mit Isobuten ist eine sanfte Methode zur Herstellung von tert.-Butylestern. Hier wird Isobuten mit der Carbonsäure in Gegenwart einer starken Säure wie Schwefelsäure umgesetzt.
- Die Reaktion von Carbonsäuren mit Diazoalkanen ist eine sehr sanfte und quantitative Methode, um Ester herzustellen. Sie wird aufgrund der schlechten Zugänglichkeit von komplexen Diazoalkanen jedoch meist nur für die Herstellung von Methyl- und Benzhydril-Estern benutzt.
Neben diesen klassischen Methoden der Veresterung wurden für spezielle Reaktionen weitere und modernere Methoden entwickelt.
- Die Aktivierung der Carbonsäure mit Dicyclohexylcarbodiimid und Umsetzung des so erhaltenen O-Acylisoharnstoffes mit der Alkoholkomponente in Gegenwart von 4-N,N-Dimethylaminopyridin (Steglich-Veresterung).
- Die Aktivierung der Carbonsäure durch Herstellung eines gemischten Anhydrides mit der 2,4,6-Trichlorbenzoesäure durch Umsetzung der Carbonsäure mit Benzoylchlorid in Gegenwart von 4-N,N-Dimethylaminopyridin und Triethylamin. Das gemischte Anhydrid wird in situ hergestellt und sofort weiter mit der Alkoholkomponente umgesetzt (Yamaguchi-Veresterung).
- Die Aktivierung der Alkoholkomponete durch die Umsetzung unter Mitsunobu-Bedingungen mit Diethylazodicarboxylat und Triphenylphosphin und anschließenden Umsetzung in situ mit der Carbonsäure (Mitsunobu-Veresterung).
Als Alkoholkomponente können verschiedene Gruppen dienen. Sehr gebräuchlich sind hier jedoch die Methyl-, die tert.-Butyl, die Benzyl- und die Allylester. Darüber hinaus kommen noch eine Reihe Schutzgruppen hinzu, welche sich aus den Ether-Schutzgruppen der Hydroxylgruppen herleiten. Die spezifischen Abspaltbedingungen sind jedoch häufig sehr ähnlich. Grundsätzlich kann jeder Ester in Anwesenheit von Hydroxidionen in wässrig-alkoholischer Lösung hydrolysiert werden. Bei empfindlicheren Substraten verwendet man jedoch häufig Lithiumhydroxid in Tetrahydrofuran und in Anwesenheit von Methanol. Für die Hydolysetendenz gelten naturgemäß die gleichen Regeln wie bei den Estern als Alkoholschutzgruppe.
| Name | Formel | Abkürzung | Spaltung | Besondere Herstellung |
|---|---|---|---|---|
| Methyl | Me | nukleophil-alkalisch durch Metallhydroxide oder Carbonate in wässrigem Methanol oder Tetrahydrofuran,[90][91] Alkalimetallhalogenide in feuchten aprotischen Lösungsmitteln wie Dimethylsulfoxid, N,N-Dimethylformamid in der Hitze,[92][93][94] Enzymatisch z. B. durch Schweineleberesterase[95][96] | Diazomethan in Diethylether,[97][98][99] Caesiumcarbonat und Methyliodid in N,N-Dimethylformamid,[100] Methanol und katalytisch Trimethylsilylchlorid[101] | |
| tert.-Butyl | |
tert.-Bu | sauer; Trifluoressigsäure (rein oder in Dichlormethan),[102] Ameisensäure, p-Toluolsulfonsäure[103] | Isobuten in Dioxan und katalytisch Schwefelsäure[104][105][106] |
| Benzyl | |
Bn | hydrogenolytisch; Wasserstoff/Palladium auf Aktivkohle[107] | |
| Benzhydryl |  |
hydrogenolytisch; Wasserstoff/Palladium auf Aktivkohle (sehr leicht zu spalten)[108] | ||
| Allyl | Allyl | analog der Ether; Kalium-tert.-Butanolat, Palladium auf Aktivkohle, DABCO (1,4-Diazabicyclo[2.2.2]octan) in Methanol, diverse Platin-Element-Komplexe – anschließend saure Aufarbeitung[109] |
Alkene Bearbeiten
Alkene werden und müssen selten durch eine Schutzgruppe geschützt werden. Sie sind in der Regel nur durch elektrophile Angriffe, Isomerisierung und bei der katalytischen Hydrierung von unerwünschten Nebenreaktionen betroffen. Grundsätzlich kennt man für Alkene zwei Schutzgruppen:
- Die temporäre Halogenierung mit Brom zur trans-1,2-Dibromalkyl-Verbindung: Die Regeneration des Alkens erfolgt unter Wiederherstellung der Konformation durch elementares Zink[110][111][112][113][114] oder mit Titanocendichlorid[115]
- Die Schützung durch eine Diels-Alder-Reaktion: Die Umsetzung eines Alkens mit einem Dien führt zu einem cyclischem Alken, welches jedoch durch elektrophile Angriffe ähnlich gefährdet ist wie das ursprüngliche Alken. Die Abspaltung des als Schutzgruppe dienenden Diens erfolgt thermisch, da es sich bei einer Diels-Alder-Reaktion um eine reversible bzw. Gleichgewichtsreaktion handelt.[116][117][118]

Alkine Bearbeiten
Für Alkine kennt man ebenfalls zwei Typen von Schutzgruppen. Bei terminalen Alkinen ist es manchmal notwendig, das acide Wasserstoffatom zu maskieren. Dies erfolgt normalerweise durch Deprotonierung (mittels starker Basen wie Methylmagnesiumbromid oder Butyllithium in Tetrahydrofuran/Dimethylsulfoxid) und anschließender Umsetzung mit Chlortrimethylsilan zum terminal TMS-geschützten Alkin. Die Abspaltung erfolgt hydrolytisch – mit Kaliumcarbonat in Methanol – oder durch Fluorid-Ionen wie beispielsweise mittels Tetrabutylammoniumfluorid.[119]

Um die Dreifachbindung selbst zu schützen, wird manchmal ein Komplex der Alkin-Verbindung mit Di-Cobaltoctacarbonyl benutzt. Die Abspaltung des Cobalts erfolgt durch Oxidation.[120][121][122][123][124]

Anwendungen Bearbeiten
Schutzgruppen finden in weiten Bereichen der organischen Synthesechemie ihre Anwendung. Dies betrifft sowohl die Laborsynthesen als auch großtechnische Synthesen von komplexen Wirkstoffen. Sobald eine funktionelle Gruppe sich als störend erweist oder aber unerwünscht angegriffen werden kann, findet die Schutzgruppentechnik ihre Anwendung. Beinahe bei jeder Synthese eines komplexen Zielmoleküls kommen Schutzgruppen zum Einsatz.[11][12] Da sowohl das Einführen als auch das Abspalten der Schutzgruppen neben dem Aufwand auch einen Ausbeuteverlust zur Folge hat, ist es erstrebenswert, ohne Schutzgruppen auszukommen, was jedoch oft nur schwer zu realisieren ist.
In der automatisierten Synthese von Peptiden und Nukleotiden ist die Schutzgruppenchemie ein integraler Bestandteil des Synthesekonzepts.[17] Aus der Zuckerchemie sind Schutzgruppen aufgrund der sehr ähnlichen Hydroxylgruppen in den Zuckermolekülen ebenfalls nicht wegzudenken.[75]
Ein wichtiges Beispiel für die industrielle Anwendung der Schutzgruppentechnik ist die Synthese von Ascorbinsäure (Vitamin C) nach Reichstein.

Um eine Oxidation der sekundären Alkohole durch Kaliumpermanganat zu verhindern, werden diese durch Acetalisierung mit Aceton geschützt und nach der Oxidation der primären Hydroxylgruppe zur Carbonsäure wieder entschützt.[125]
Ein sehr spektakuläres Beispiel aus der Naturstoffsynthese zur Anwendung von Schutzgruppen ist die Totalsynthese von Palytoxin-Carbonsäure durch die Arbeitsgruppe um Yoshito Kishi aus dem Jahre 1994.[126] Hier mussten 42 Funktionelle Gruppen (39 Hydroxylgruppen, ein Diol, eine Aminogruppe und eine Carbonsäuregruppe) geschützt werden. Dies erfolgte durch acht verschiedene Schutzgruppen (ein Methylester, fünf Acetat-Gruppen, 20 TBDMS-Ether, neun p-Methoxybenzylether, vier Benzoate, ein Methyl-halbacetal, ein Acetal mit Aceton und ein SEM-Ester.[127]

Leider beeinflusst das Einführen oder Verändern einer Schutzgruppe bisweilen auch die Reaktivität des Gesamtmoleküls. Als Beispiel sei hier ein Ausschnitt aus der Synthese eines Analogons vom Mitomycin von Danishefsky gezeigt.[128]

Der Wechsel der Schutzgruppe von einem Methylether zu einem MOM-Ether verhindert hier das Öffnen des Epoxides zum Aldehyd.
Eine bedeutende Anwendung der Schutzgruppenchemie findet sich in der automatisierten Synthese von Peptiden und Nucleosiden. Bei der Peptidsynthese durch automatische Synthesiser wird die Orthogonalität der Fmoc-Gruppe (basische Spaltung), der tert.-Butyl-Gruppe (saure Spaltung) und diverse Schutzgruppen für funktionelle Gruppen in der Seitenkette der Aminosäuren ausgenutzt.[17] Bei der automatisierten Nukleotid-Synthese von DNA– und RNA-Sequenzen werden Schutzgruppen zum einen zum Blockieren von funktionellen Gruppen verwendet aber es erfolgt auch Redox-Chemie an den geschützten Atomen. Der Phosphor wird geschützt und während des Kupplungszyklusses zum Phosphat oxidiert.[129]

In der Regel ist das Einführen einer Schutzgruppe unproblematisch. Die Schwierigkeiten liegen eher in ihrer Stabilität und beim selektiven Abspalten. Leider werden auftretende Probleme bei Synthesestrategien mit Schutzgruppen nur selten in der Fachliteratur dokumentiert.[130]
Atomökonomie Bearbeiten
Synthesen unter Einsatz von Schutzgruppen weisen in der Regel eine geringe Atomökonomie auf.[131][132][133] Manchmal muss der Umweg der Verwendung von Schutzgruppen eingeschlagen werden, um unerwünschte Konkurrenzreaktionen auszuschalten und die angestrebte Selektivität einer Synthese zu erreichen.[134] Bei der Synthese komplexer Strukturen sind Schutzgruppenstrategien oft unverzichtbar.
Als Beispiel für eine Schutzgruppenstrategie im Vergleich zu einer schutzgruppenfreien Synthese seien die Synthesen von Hapalindole U gegenübergestellt. Während die Synthese von Hideaki Muratake aus dem Jahr 1990 Tosyl als Schutzgruppe verwendet,[135][136][137] wurde in der Synthese von Phil S. Baran aus dem Jahre 2007 auf jede Schutzgruppe verzichtet.[138] Dabei wurde die Zahl der Syntheseschritte maßgeblich verringert.
-
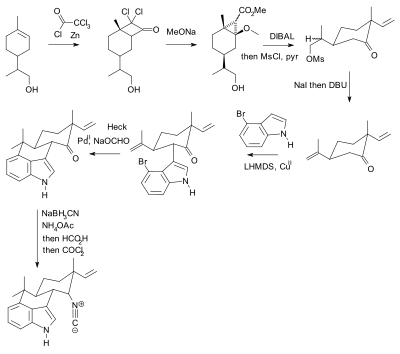 Hapalindole U Baran 2007 Schutzguppenfreie Synthese
Hapalindole U Baran 2007 Schutzguppenfreie Synthese -
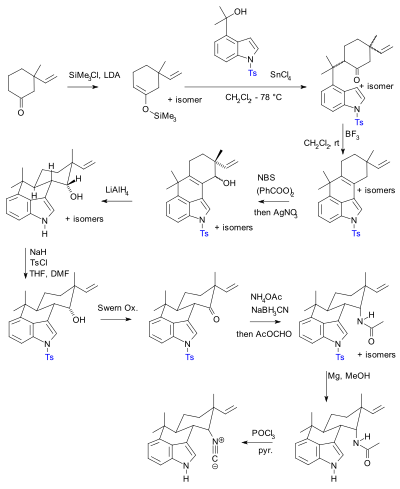 Hapalindole U Muratake 1990 Ts Schutzgruppensynthese (Schutzgruppen in blau)
Hapalindole U Muratake 1990 Ts Schutzgruppensynthese (Schutzgruppen in blau)


Literatur Bearbeiten
- Phillip J. Kocieński: Protecting Groups, 1. Auflage, Georg Thieme Verlag, Stuttgart 1994, ISBN 3-13-135601-4.
- Peter G.M. Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis, 4th Ed., John Wiley & Sons Inc., Hoboken, New Jersey, ISBN 0-471-69754-0.
- Michael Schelhaas, Herbert Waldmann: „Schutzgruppenstrategien in der organischen Synthese“, in: Angewandte Chemie, 1996, 103, S. 2192–2219; doi:10.1002/ange.19961081805.
- Krzysztof Jarowicki, Philip Kocienski: „Protecting groups“, in: J. Chem. Soc., Perkin Trans. 1, 1998, S. 4005–4037; doi:10.1039/A803688H.
Weblinks Bearbeiten
- Universität Marburg: Schutzgruppen in der organischen Synthesechemie
- Organic-Reaction.com: Protecting Group
- Organic-Chemistry.org: Protecting Groups
Voltammetrie Bearbeiten
Die Voltammetrie ist eine Sammelbezeichnung für verschiedene elektroanalytische Methoden zur qualitativen und quantitativen Analyse einer Probe, mit der man die chemische Zusammensetzung von Stoffgemischen anhand des spannungsabhängigen Stromverlaufs bestimmen kann, sowie zur Aufklärung von Reaktionsmechanismen.
Das Wort Voltammetrie ist ein Kunstwort aus Volt- und Amperometrie und zeigt auf, dass die Grundlage der Voltammetrie Strom-Spannungs-Kurven sind. Bei der Messung der Stromstärke zwischen zwei Festkörperelektroden wird die Spannung mit der Zeit variiert. (Im Gegensatz dazu wird bei der Voltametrie (mit einem „m“) die Spannung U gemessen.)
Dieses Verfahren ist eine Form der Elektrolyse in Flüssigkeiten oder Gasen, bei der man findet, dass spezielle chemische Bestandteile besonders bei einer ihnen typischen Spannung durch die sich bildende Stoffverteilung beschleunigt werden. Die Durchtrittsreaktion führt zu einem plötzlichen Stromanstieg, wenn das elektrische Feld ausreicht, um die Moleküle, Atome oder Ionen innerhalb der Doppelschicht zu oxidieren oder zu reduzieren. Kennzeichen dieser Reaktion ist ein Durchtritt von Elektronen durch die Grenzfläche zwischen Elektronenleiter und Elektrolyt.
Die Auswertung von Stromspannungskurven stellt eine in der Galvanotechnik, vor allem bei der alkalischen Verzinkung und sauren Verkupferung, bevorzugte Methode zur Bewertung der Abscheidungsbedingungen dar. Hier werden nicht nur - wie oben dargelegt - einzelne Abschnitte, sondern die gesamte Kurve als Summenparameter ausgewertet.
Inversvoltammetrie Bearbeiten
Bei dieser Form der Voltammetrie wird vor dem Potentialscan ein Anreicherungsschritt durchgeführt. Ziel ist es, durch Abscheidung des Analyten an der Arbeitselektrode größere voltammetrische Signale zu erhalten, um quantitative Bestimmungen im Ultraspurenbereich zu ermöglichen. Dafür gibt es unterschiedliche Möglichkeiten. Durch Elektrolyse können Metallionen zum Metall reduziert und als hauchdünne Schicht an der Arbeitselektrode abgeschieden werden. Im Falle von Quecksilber als Elektrodenmaterial (hängender Tropfen oder Film) wird dabei meist eine Legierung gebildet. Das Metall löst sich im flüssigen Quecksilber. Insbesondere Metallkomplexverbindungen und organische Analyten lassen sich durch Adsorption an der Arbeitselektrode ablagern. Die Voranreicherung des Analyten bewirkt eine Absenkung der Bestimmungsgrenze um den Faktor 1000.
Beispiel:
Cadmiumionen werden im Anreicherungsschritt an einer Quecksilberelektrode abgeschieden:

In Umkehrung, dem sogenannten Stripping, wird das abgeschiedene Cadmium anodisch wieder oxidiert:

Hierbei wird die Strom-Spannungskurve aufgenommen, wobei bei linear veränderlicher Spannung Spitzenströme auftreten, die registriert werden. Über das aufgenommene Diagramm lassen sich Aussagen über die ursprüngliche Konzentration des Metallions in der Lösung machen, wenn man zuvor Eichkurven erstellt hat. Man kann bei dem umgekehrten Vorgang auch andere Bestimmungsverfahren, z.B. die Coulometrie anwenden, wobei in diesem Fall über die ermittelte Ladung die Masse des zuvor abgeschiedenen Stoffes berechnet werden kann.
Liegen mehrere Metallionen in der Lösung vor, so entstehen bei der anodischen Wiederauflösung mehrere Stromspitzen, die man entsprechend zuvor erstellter Eichkurven den unterschiedlichen Metallionen zuordnen kann.
Siehe auch Bearbeiten
Polarographie, Amperometrie, Konduktometrie, Elektrochemie, Redoxsystem, Freie Energie, Zyklovoltammetrie
Literatur Bearbeiten
- Rolf Neeb: Inverse Polarographie und Voltammetrie, Angew. Chem., 74. Jahrg. 1962, S. 203
Weblinks Bearbeiten
Coulometrie Bearbeiten
Die Coulometrie ist eine Methode der Elektrochemie.
Die Coulometrie ist eine Methode, um die quantitative Stoffmenge einer oxidierbaren oder reduzierbaren Verbindung zu ermitteln. Sie wurde von den Ungarn Szebelledy und Somgyi im Jahre 1938 entwickelt. Erst ab 1950 fand dann die Methode breitere Anwendung. Die Coulometrie beruht auf der Messung der elektrischen Ladung bzw. Elektrizitätsmenge, die an einer Arbeitselektrode umgesetzt wird. Die Coulometrie ist der Elektrogravimetrie sehr ähnlich, jedoch werden hierbei die oxidierten oder reduzierten Stoffe nicht an der Elektrode abgeschieden sondern bleiben in Lösung und die vollständige Umsetzung kann nur über einen indirekten Indikator (z. B. Manganation) angezeigt werden. Gemäß dem Faradayschen Gesetz ist die elektrische Ladung proportional zur umgesetzten Stoffmenge. Bei vollständigem elektrochemischen Umsatz des zu bestimmenden Stoffes (des Analyten) und fehlenden elektrochemischen Nebenreaktionen kann mittels der Faradaykonstante die Analytmenge ausgerechnet werden. Da an der Gegenelektrode ebenfalls eine elektrochemische Reaktion ablaufen muss, um den Stromkreis zu schließen, muss gewährleistet werden, dass die Reaktionsprodukte nicht in den Bereich der Arbeitselektrode gelangen können. Das kann durch ein Diaphragma oder mittels chemischer Bindung (z. B. Halogen mit Silber-Gegenelektrode als schwer lösliches Silberhalogenid) geschehen. Die Coulometrie findet z. B. Anwendung bei der Bestimmung des Wassergehaltes nach Karl Fischer im Spurenbereich oder bei der Quantifizierung der adsorbierbaren organisch gebundenen Halogenen (AOX) in Wasserproben.
Potentiostatische Coulometrie Bearbeiten
Bei der potentiostatischen Variante der Coulometrie wird das Elektrodenpotential mit Hilfe eines Potentiostaten konstant gehalten. Diese Potentialkontrolle ist sehr vorteilhaft, um Nebenreaktionen auszuschließen. Von Nachteil ist, dass die Stromstärke wegen der stetig sinkenden Analytkonzentration stark abfällt. Dadurch kann das Experiment viel Zeit in Anspruch nehmen. Das Ende der Reaktion wird angenommen, wenn der Stromabfall 99,9 Prozent erreicht hat. Die gesuchte Elektrizitätsmenge errechnet sich dann nach dem Integral des Stromes über die Zeit. Bei sehr kleiner Elektrodenoberfläche, großen Konzentrationen und großen Volumina kann die Analyse auch Tage in Anspruch nehmen. Aus diesem Grund ist die Coulometrie im ausgesprochenem Maße eine spurenanalytische Methode.

Diese Auswertung übernimmt meist eine integrierende analoge Schaltung (Integratorschaltung) oder ein Computerprogramm.
Die Berechnung der abgeschiedenen oder umgesetzten Masse ergibt sich aus folgenden Beziehungen:
Faraday-Gesetz:
Stoffmenge:
Eingesetzt und aufgelöst nach der Masse m gilt:
Hierbei ist M die molare Masse in g/mol, Q die experimentell bestimmte Ladung in Coulomb, z die Ladungszahl, die bei Reduktion bzw. Oxidation der Änderung der Oxidationszahl entspricht und F die Faraday-Konstante in C/mol.



Beispiele Bearbeiten
Reduktion von Metallionen zu Metallen an Quecksilber- oder Platinelektroden:

Änderung der Oxidationsstufe (Wertigkeitsstufe) an Platinelektroden:

Oxidative Abscheidung von Halogeniden an Silberelektroden:

Galvanostatische Coulometrie Bearbeiten
Bei der galvanostatischen Variante der Coulometrie wird die Elektrolyse-Stromstärke mit Hilfe eines Galvanostaten konstant gehalten. Dieser besteht im einfachsten Falle aus einer Batterie, einem Widerstand von etlichen Kiloohm und einem Potentiometer in Reihenschaltung mit der elektrochemischen Zelle. Der Kiloohm-Widerstand begrenzt den elektrischen Strom, da er den weitaus höchsten Widerstandswert im Stromkreis besitzt. Von Vorteil sind die einfache Gerätetechnik und die schnelle Durchführung. Nachteilig wirkt sich aus, dass das Elektrodenpotential sich während der Reaktion verändert und somit Nebenreaktionen durch andere Maßnahmen (z. B. Reinigungsschritte bei der Probevorbereitung) ausgeschlossen werden müssen. Das Ende der Reaktion muss durch eine Indikationsmethode angezeigt werden (beispielsweise durch eine pH-Wert-Messung). Diese Methode kann daher auch als eine "Titration mit Elektronen" angesehen werden.
Da die Stromstärke konstant gehalten wird, gilt für die umgesetzte Ladung folgende Beziehung:

Indikationsmethoden Bearbeiten
Die untenstehenden Methoden können zur Endpunktsindizierung genutzt werden. Dabei ist zu beachten, dass die Güte der Methode oftmals von der Analytmenge abhängig ist und von der Hintergrundmatrix, wie z.B. pH-Puffer etc.
pH-Indikation: Die pH-Indikation ist bei pH=7 am besten; je weiter man zu pH=0 oder pH=14 wandert, desto schlechter wird die Indikation. Bei sehr geringer Analytkonzentration ist die Änderung des pH-Werts nicht groß genug, um eine pH-Wert Änderung festzustellen, da Wasser auch Puffereigenschaften besitzt.
Konduktometrische Indikation: Bei Reaktionen, die nicht zwischen pH=6 und pH=8 verlaufen, ist die Leitfähigkeit der Protonen oder der Hydroxidionen zu groß. Außerdem wird immer Leitsalz im Überschuss zugegeben, um Migration des Analyten im elektrischen Feld zu verhindern. Daher ist die Leitfähigkeit der Lösung generell hoch. Bei geringer Analytkonzentrationen wird die Leitfähigkeit nicht signifikant geändert. Somit ist eine Indikation schwierig bis unmöglich, solange die Analytkonzentration gering ist.
Photometrische Indikation: Bei sehr geringen Ausgangskonzentration ist der Extinktionskoeffizient der meisten Analyten zu gering, um eine signifikante Änderung der Extinktion wahrzunehmen. Durch die hohe Konzentration an Leitsalz und Hilfsreagenz, können matrixbedingte Störungen auftreten.
Biamperometre Indikation mittels einer Indikatorelektrode: Man legt an einer Indikatorelektrode ein sehr kleines Potential oder einen sehr kleinen Strom an und gibt ein Hilfsreagenz in die Lösung, das umgesetzt wird, anstelle vom Analyten. Da man für den ungehinderten Stromfluss eine Reaktion an der Kathode und an der Anode zulassen muss und das gelöste Salz nur in einer Oxidationsstufe vorliegt (oxidiert oder reduziert), fließt nur ein kleiner Reststrom. Sobald man mit der Coulometrie beginnt, wird das Hilfsreagenz umgesetzt, das danach den Analyten umsetzt und zurückreagiert. Die für die Oxidation/Reduktion wichtige Spezies wird wieder umgesetzt, der Stromfluss bleibt also sehr klein. Nach dem Umsatz des Analyten wird die oxidierte und die reduzierte Form in der Lösung durch die Reaktion vorliegen. Dadurch kann an der Indikatorelektrode (die meist aus zwei Pt-Stiften besteht) der Strom fließen, da jetzt an der Anode und an der Kathode die elektrochemischen Prozesse ablaufen können. Legt man einen definierten Strom an, fällt das Potential ab, legt man ein festes Potential an, steigt der Strom nach der vollständigen Umsetzung des Analyten. Da bei der Galvanostatischen Coulometrie eine Potentialerhöhung mit der Zeit auftritt, muss sowieso ein Hilfsreagenz im Überschuss eingesetzt werden, die die Potentialerhöhung vernachlässigbar klein werden lässt. Dieses Hilfsreagenz kann dann leicht zur Endpunktsbestimmung herangezogen werden. Glücklicherweise ist der Spannungsabfall bzw. die Stromerhöhung in der Lösung nicht von der Analytkonzentration abhängig, sondern von der Indikatorelektrodenoberfläche (möglichst klein halten) und der Konzentration des Hilfsreagenz.
Beispiel: Bearbeiten
Es sollen Cer(IV)-ionen bestimmt werden. Hierbei werden diese Ionen bei der Bestimmung reduziert:

Die eigentliche Reduktion erfolgt dadurch, dass ein im Überschuss zugegebenes Hilfsreagenz (z.B. ein Eisen(III)-salz) bei der Elektrolyse kathodisch reduziert wird und die reduzierte Form dann bei der Oxidation die Elektronen liefert:


Solange nicht alle Cer(IV)-ionen reduziert worden sind, bleibt die Konzentration der Eisen(III)-ionen konstant und es fließt ein konstanter Strom. Der Endpunkt der coulometrischen Bestimmung ist dann erreicht, wenn die Stromstärke sich verringert.
Chronocoulometrie Bearbeiten
Bei der Chronocoulometrie handelt es sich im Grunde um eine Chronoamperometrie, d. h. es wird ein Potentialsprungexperiment durchgeführt und die Änderung des Elektrolysestroms mit hoher zeitlicher Auflösung (Mikrosekunden) verfolgt. Allerdings wird über die Zeit integriert, um die umgesetzte elektrische Ladung zu erhalten. Das Ziel besteht in der Bestimmung oberflächlich an der Arbeitselektrode befindlicher Stoffe. Diese werden in kürzester Zeit umgesetzt, während im Elektrolyten gelöste Substanzen erst zur Elektrodenoberfläche diffundieren müssen. Der elektrische Strom, der durch letzteren Vorgang verursacht wird, fällt gemäß der Cottrellgleichung mit ab. Daher kann rechnerisch zwischen der Umsetzung gelöster und abgeschiedener Stoffe unterschieden werden.

Siehe auch Bearbeiten
Literatur Bearbeiten
- Abresch/Büchel: Die coulometrische Analyse, Angew. Chem. 74. Jahrg. 1962, Nr. 17, S. 685
- Georg Schwedt: Analytische Chemie, Wiley VCH Verlagsgesellschaft, 2. Auflage 2008, S. 175 ff., ISBN 978-3-527-31206-1
Amperometrie Bearbeiten
Die Amperometrie ist eine elektrochemische Methode zur quantitativen Bestimmung von chemischen Stoffen. Bei der amperometrischen Titration wird der elektrochemisch erzeugte Stromfluss als Nachweis für die Vollständigkeit einer Umsetzung genutzt.
Allgemeines Bearbeiten
Die amperometrische Methode ist gekennzeichnet durch die Messung eines Elektrolysestroms an einer Arbeitselektrode, während ein zeitlich konstantes elektrochemisches Potential anliegt. Damit leitet sich die Amperometrie von der Voltammetrie ab, bei welcher die Elektrolysespannung mit der Zeit verändert wird. Bei einer Titration muss entweder die vorliegende Lösung oder die zutitrierte Lösung Stoffe enthalten, die an den Elektroden oxidiert oder reduziert werden können. Man misst die Stromstärke in Abhängigkeit von der zugesetzten Lösungsmenge. Ist die Zugabe bei der Titration vollständig, kann der Strom ansteigen oder abfallen (je nach Redoxsystem).
Der gemessene Elektrolysestrom ist der Konzentration des umgesetzten Stoffes direkt proportional. Dies gestattet eine Bestimmung unbekannter Konzentrationen mit Hilfe einer Kalibrierfunktion. Häufig verwendete Materialien für Arbeitselektroden sind: Platin, Gold, Kohlenstoff, Quecksilber und Silber. Beim Clark-Sensor wird gelöster Sauerstoff bei einem konstanten Potential reduziert. Dieses Prinzip der Sauerstoffbestimmung ist vielfach in Industrie und Umweltanalytik Gebrauch. Die Arbeitselektroden amperometrischer Sensoren können mit einer Schicht überzogen sein, die selektiv mit dem zu analysierenden Stoff reagiert. Sehr weit verbreitet in der medizinischen Diagnostik sind amperometrische Glucosesensoren, die mit dem Enzym Glucose-Oxidase modifiziert sind. In seiner Funktion als Biokatalysator setzt dieses Enzym den Analyten Traubenzucker (Glucose) zu Gluconsäure und Wasserstoffperoxid um. Dabei wird Sauerstoff verbraucht. Amperometrisch registriert wird eigentlich die Zunahme der Wasserstoffperoxidkonzentration oder die Abnahme der Sauerstoffkonzentration, je nach Wahl des Elektrolysepotentials.
Chronoamperometrie Bearbeiten
Hierbei handelt es sich um eine Relaxationsmethode mit einem Potentialsprung und Registrierung des sich ändernden Elektrolysestroms. Zunächst wird ein Potential an die Arbeitselektrode angelegt, bei welchem noch kein Umsatz des Analyten erfolgt. Durch sprunghafte Änderung des Potentials auf einen neuen zeitlich konstanten Wert beginnt die Oxidation bzw. Reduktion des Analyten und ein elektrochemischer Strom beginnt zu fließen. Dieser Strom hat unmittelbar nach dem Potentialsprung seinen maximalen Wert und fällt dann ab. Der zeitliche Verlauf wird durch die Cotrellgleichung beschrieben.

Hierin bedeuten:
- I - Elektrolysestrom
- z - Zahl der übertragenen Elektronen
- F - Faraday-Konstante (96.486 As/mol)
- D - Diffusionskonstante (u. a. abhängig von der Viskosität der Lösung und der Größe der diffundierenden Teilchen)
- A - Elektrodenoberfläche
- t - Zeit
- c - Ausgangskonzentration des umgesetzten Stoffes
Das Produkt ist dabei für den untersuchten Stoff in einem bestimmten Zeitraum während der Messung konstant und ist abhängig von der Ausgangskonzentration c, der Diffusionskonstanten D und der Zahl der übertragenen Elektronen z (Änderung der Oxidationsstufe des Stoffes). Hierüber lassen sich demnach über die Cotrell-Gleichung Berechnungen der Ausgangskonzentration oder der Änderung der Oxidationsstufe oder der Diffusionskonstanten durchführen.[139]

Biamperometrie Bearbeiten
Bei dieser vereinfachten Variante der Amperometrie wird mit zwei gleichen Arbeitselektroden gearbeitet, die beispielsweise aus je einem Platindraht bestehen. Zwischen den beiden Elektroden liegt ein kleiner Widerstand (z. B. 10 Ohm), zwischen der Anodenelektrode und dem Pluspol liegt ein großer Widerstand (z. B. 4 Kilo Ohm), bei Batteriespannungen von 0,5 - 1 V wird der Strom an der Anode gemessen. Es kann nur dann ein Elektrolysestrom fließen, wenn an beiden Elektroden Stoffumsetzung erfolgt. Dies tritt dann ein, wenn beide Komponenten elektroaktive Spezies sind und die elektrische Spannung zwischen beiden Elektroden hinreichend groß ist (meist 10-100 mV). Verwendet wird diese Methode bei der Dead-Stop-Titration.
Die Dead-Stop-Titration wird beispielsweise zum Nachweis geringer Wasserspuren mittels der Karl-Fischer-Titration gebraucht. Dabei setzt sich Schwefeldioxid mit Iod und Wasser zu Iodid und Schwefelsäure um. Vor dem Endpunkt liegt noch das inaktive Iodid vor. Sobald aber freie Iodmoleküle vorliegen kommt es zu einem Stromanstieg.
Siehe auch: Bearbeiten
Literatur Bearbeiten
- Ullmanns Enzyklopädie der technischen Chemie, 4. Auflage, Stichwort: Elektrochemische Analyseverfahren.
- Georg Schwedt, Analytische Chemie, Wiley VCH, 2. Auflage 2008, ISBN 978-3-527-31206-1
- Karl Cammann (Hrsg.), Instrumentelle Analytische Chemie, Spektrum Akademischer Verlag, Heidelberg - Berlin, 2001.
Derivat (Chemie) Bearbeiten
Als Derivat (auch Abkömmling, lateinisch derivare, abfließen, ableiten) wird in der Chemie ein abgeleiteter Stoff ähnlicher Struktur zu einer entsprechenden Grundsubstanz bezeichnet. Derivate sind Stoffe, deren Moleküle an Stelle eines H-Atoms oder einer funktionellen Gruppe ein anderes Atom oder eine andere Atomgruppe besitzen bzw. bei denen ein oder mehrere Atome/Atomgruppen entfernt wurden. Die Herstellung eines Derivates bezeichnet man als Derivatisierung.
Beispiele Bearbeiten
Man bezeichnet Carbonsäureamide, Carbonsäureanhydride, Carbonsäurehalogenide und Nitrile als Derivate der jeweiligen Carbonsäure, da sie aus dieser durch Veränderung funktioneller Gruppen zugänglich sind. Alkohole und Ether kann man formal als Derivate des Wassers mit der Struktur
- R-O-H bzw. R-O-R
ansehen. Setzt man für den Rest R Wasserstoff, so erhält man Wasser (H-O-H), setzt man für R den Methylrest CH3, so erhält man Methanol (CH3-O-H).
Derivate des Ethans sind beispielsweise
- Ethanol
- Ethen
- Ethin
- Acetaldehyd (Ethanal)
- Essigsäure (Ethansäure)
- Chlorethan
- Ethylamin (Aminoethan)
Homologe Reihen Bearbeiten
Vom Begriff Derivat abgegrenzt werden muss der Begriff Homologon. Homologa sind Stoffe, die sich nur durch die Kettenlänge ihrer Grundbausteine unterscheiden; in der Organischen Chemie sind dies die Kohlenwasserstoffketten etwa von Alkanen, Alkenen, Alkoholen oder Carbonsäuren. Homologa sind in aller Regel keine Derivate.
Die Bedeutung von Derivaten Bearbeiten
In der Pharmazie hat die Derivatisierung besondere Bedeutung, um aus bestehenden Arzneimitteln neue, möglicherweise wirksamere oder verträglichere Arzneimittel zu schaffen. So ist zum Beispiel die Acetylsalicylsäure (der Hauptbestandteil und -wirkstoff des Aspirins) ein Derivat der Salicylsäure, also des medizinisch aktiven Stoffes. Ein weiteres Beispiel ist Paracetamol, das besser verträgliche Derivat des Acetanilids.
In der gesamten chromatographischen Analytik spielen Derivate eine bedeutende Rolle. In der Gaschromatographie und Gaschromatographie mit Massenspektrometrie-Kopplung werden Derivate meist eingesetzt, um nicht oder nur schwer verdampfbare Analyte in leichter flüchtige Derivate umzuwandeln, die der Chromatographie in der Gasphase zugänglich sind. In der HPLC werden häufig chromophore und/oder fluorophore Derivate hergestellt, um die sensitive und spezifische Detektion im sichtbaren bzw. Ultraviolett-Bereich oder durch den Nachweis mittels Fluoreszenzdetektoren zu ermöglichen.
Literatur Bearbeiten
- Karl Blau, Graham S. King (Hrsg.): Handbook of Derivatives for Chromatography. Heyden & Son Ltd., London 1977, ISBN 0-85501-206-4.
- Jürgen Falbe u. a. (Hrsg.): Römpp Lexikon Chemie. 9. Aufl. Thieme, Stuttgart 1995, ISBN 3-13-102759-2.
- Daniel R. Knapp: Handbook of Analytical Derivatization Reactions. J. Wiley & Sons, New York 1997, ISBN 0-471-03469-X.
- Kurt Peter C. Vollhardt: Organische Chemie („Organic chemistry, structure and function“). 3. Aufl. Wiley VCH, Weinheim 2000, ISBN 3-527-29819-3 (Kap. 15, 19 und 20).
@@@@@
Periodensystem Bearbeiten
Atom Bearbeiten
Chemische Bindung Bearbeiten
Molekül Bearbeiten
Komplexchemie Bearbeiten
(Wikibook: Ligandenfeldtheorie und die Vereinheitlichung molekularen Verhaltens)
Chemische Reaktion Bearbeiten
Reaktionsgleichung Bearbeiten
Stöchiometrie Bearbeiten
Thermodynamik Bearbeiten
Chemisches Gleichgewicht Bearbeiten
Massenwirkungsgesetz Bearbeiten
Dissoziation (Chemie) Bearbeiten
Elektrische Leitfähigkeit Bearbeiten
Elektrochemie Bearbeiten
Elektrode Bearbeiten
Nernst-Gleichung Bearbeiten
Elektrochemische Spannungsreihe Bearbeiten
pH-Wert Bearbeiten
Kinetik (Chemie) Bearbeiten
Lesenswerte Artikel Bearbeiten
Einzelnachweise Bearbeiten
- ↑ a b Walter Wittenberger: Chemische Laboratoriumstechnik, Springer-Verlag, Wien, New York, 7. Auflage, 1973, S. 209−211, ISBN 3-211-81116-8.
- ↑ Joseph C. Touchstone: Practice of Thin Layer Chromatography. 3. Auflage, Wiley, New York, 1992, S. 3–4.
- ↑ Ullmanns Encyklopädie der technischen Chemie. 4. Auflage. Band 5, S. 193.
- ↑ K. Günther, J. Martens, M. Schickedanz: Dünnschichtchromatographische Enantiomerentrennung mittels Ligandenaustausch. In: Angew. Chem.. 96, 1984, S. 514–515, DOI: 10.1002/ange.19840960724.
- ↑ K. Günther: Thin-layer chromatographic enantiomeric resolution via ligand exchange. In: J. Chrom. 448, 1988, S. 11–30.
- ↑ K. Günther, M. Schickedanz, J. Martens: Thin-Layer Chromatographic Enantiomeric Resolution. In: Naturwissenschaften. 72, 1985, S. 149–150.
- ↑ Teresa Kowalska, Joseph Sherma (Hrsg.): Thin Layer Chromatography in Chiral Separations and Analysis. CRC Press Taylor & Francis Group, 2007, ISBN 978-0-8493-4369-8 (Chromatographic Science Series. Band 98).
- ↑ Mitchell E. Johnson: Rapid, Simple Quantitation in Thin-Layer Chromatography Using a Flatbeted Scanner. In: Journal of Chemical Education. Vol. 77, März 2000, S. 368.
- ↑ P. Abu-Rabie, N. Spooner: Direct Quantitative Bioanalysis of Drugs in Dried Blood Spot Samples Using a Thin-Layer Chromatography Mass Spectrometer Interface. In: Anal. Chem. 81, 2009, S. 10275–10284, PMID 19919036.
- ↑ Joseph Sherma, Bernard Fried: Handbook of thin-layer chromatography. Marcel Dekker, New York 2003, S. 323 (Eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ a b Kyriacos C. Nicolaou, Erik J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, 1996, ISBN 3-527-29284-5.
- ↑ a b c Kyriacos C. Nicolaou, Scott A. Snyder: Classics in Total Synthesis II, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003, ISBN 3-527-30684-6.
- ↑ Philipp J. Kocieński: Protecting Groups, 1. Auflage, Georg Thieme Verlag, Stuttgart 1994, ISBN 3-13-135601-4.
- ↑ Peter G.M. Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis, Fourth Ed. John Wiley & Sons Inc., Hoboken, New Jersey, ISBN 0-471-69754-0.
- ↑ P.J. Kocieński: Protecting Groups, S. 245–250.
- ↑ Dietrich Spitzner, Kai Oesterreich: „Anionically Induced Domino Reactions – Synthesis of a Norpatchoulenol-Type Terpene“, in: European Journal of Organic Chemistry, 2001, 10; S. 1883–1886; doi:10.1002/1099-0690(200105)2001:10<1883::AID-EJOC1883>3.0.CO;2-M.
- ↑ a b c Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis. Reprint 2004, Oxford University Press, ISBN 0-19-963724-5.
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis, S. 10–12.
- ↑ Kyriacos C. Nicolaou, Eric J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, VCH Verlagsgesellschaft mbH, Weinheim, 1996, S. 711–729, ISBN 3-527-29284-5.
- ↑ Michael Schelhaas, Herbert Waldmann: „Schutzgruppenstrategien in der organischen Synthese“, in: Angewandte Chemie, 1996, 103, S. 2195–2200; doi:10.1002/ange.19961081805.
- ↑ V.N. Rajasekharan Pillai: „Photoremovable Protecting Groups in Organic Synthesis“, in: Synthesis, 1980, S. 1–26.
- ↑ P.J. Kocieński: Protecting Groups, S. 186.
- ↑ Naomi Sakai, Yasufumi Ohfune: „Total synthesis of galantin I. Acid-catalyzed cyclization of galantinic acid“, in: J. Am. Chem. Soc., 1992, 114, S. 998–1010; doi:10.1021/ja00029a031.
- ↑ Glenn L. Stahl, Roderich Walter, Clarck W. Smith: „General procedure for the synthesis of mono-N-acylated 1,6-diaminohexanes“, in: J. Org. Chem., 1978, 43, S. 2285–2286; doi:10.1021/jo00405a045.
- ↑ R.A. Houghton, A. Beckman, J.M. Ostresh: Int. J. Pept. Protein Res., 1986, 27, S. 653.
- ↑ P.J. Kocieński: Protecting Groups, S. 195.
- ↑ Robert M. Williams, Peter J. Sinclir, Dongguan Zhai, Daimo Chen: „Practical asymmetric syntheses of α-amino acids through carbon-carbon bond constructions on electrophilic glycine templates“, in: J. Am. Chem. Soc., 1988, 110, S. 1547–1557; doi:10.1021/ja00213a031.
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis, S. 27–30.
- ↑ P.J. Kocieński: Protecting Groups, S. 199–201.
- ↑ John O. Osby, Michael G. Martin, Bruce Ganem: „An Exceptionally Mild Deprotection of Phthalimides“, in: Tetrahedron Lett., 1984, 25, S. 2093–2096; doi:10.1016/S0040-4039(01)81169-2.
- ↑ P.J. Kocieński: Protecting Groups, S. 220–227.
- ↑ P. Vouros: Chemical derivatization in gas chromatographie-mass spectrometrie. In: Mass Spectrometrie. Degger, New York, 1979, Bd. 2, S. 129.
- ↑ P.J. Kocieński: Protecting Groups, S. 29.
- ↑ P.J. Kocieński: Protecting Groups, S. 31.
- ↑ Tod K Jones, Robert A. Reamer, Richard Desmond, Sander G. Mills: „Chemistry of tricarbonyl hemiketals and application of Evans technology to the total synthesis of the immunosuppressant (−)-FK-506“, in: J. Am. Chem. Soc., 1990, 112, S. 2998–3017; doi:10.1021/ja00164a023.
- ↑ Dieter Seebach, Hak-Fun Chow, Richard F.W. Jackson, Marius A. Sutter, Suvit Thaisrivongs, Jürg Zimmermann: „(+)-11,11′-Di-O-methylelaiophylidene – preparation from elaiophylin and total synthesis from (R)-3-hydroxybutyrate and (S)-malate“, in: Liebigs Ann. Chem., 1986, S. 1281–1308; doi:10.1002/jlac.198619860714.
- ↑ David A. Evans, Stephen W. Kaldor, Todd K. Jones, Jon Clardy, Thomas J. Stout: „Total synthesis of the macrolide antibiotic cytovaricin“, in: J. Am. Chem. Soc., 1990, 112, S. 7001–7031; doi:10.1021/ja00175a038.
- ↑ James A. Marshall, Richard Sedrani: „A convergent, highly stereoselective synthesis of a C-11-C-21 subunit of the macbecins“, in: J. Org. Chem., 1991, 56, S. 5496–5498; doi:10.1021/jo00019a004.
- ↑ a b James D. White, Motoji Kawasaki: „Total synthesis of (+)-latrunculin A“, in: J. Am. Chem. Soc., 1990, 112, S. 4991–4993; doi:10.1021/ja00168a071.
- ↑ Morris J. Robins, Vicente Samano, Mark D. Johnson: „Nucleic acid-related compounds. 58. Periodinane oxidation, selective primary deprotection, and remarkably stereoselective reduction of tert-butyldimethylsilyl-protected ribonucleosides. Synthesis of 9-(β-D-xylofuranosyl)adenine or 3'-deuterioadenosine from adenosine“, in: J. Org. Chem., 1990, 55, S. 410–412; doi:10.1021/jo00289a004.
- ↑ R. Roger F. Newton, Derek P. Reynolds, Colin F. Webb, Stanley M. Roberts: „A short and efficient total synthesis of (±) prostaglandin D2 methyl ester involving a new method for the cleavage of a dimethyl-t-butylsilyl ether“, in: J. Chem. Soc., Perkin Trans. 1, 1981, S. 2055–2058; doi:10.1039/P19810002055.
- ↑ Kyriacos C. Nicolaou, R. A. Daines, T. K. Chakraborty: „Total synthesis of amphoteronolide B“, in: J. Am. Chem. Soc., 1987, 109, S. 2208–2210; doi:10.1021/ja00241a063.
- ↑ Leo A. Paquette, Annette M. Doherty, Christopher M. Rayner: „Total synthesis of furanocembranolides. 1. Stereocontrolled preparation of key heterocyclic building blocks and assembly of a complete seco-pseudopterane framework“, in: J. Am. Chem. Soc., 1991, 109, S. 3910–3926; doi:10.1021/ja00036a045.
- ↑ P.J. Kocieński: Protecting Groups, S. 40.
- ↑ P.J. Kocieński: Protecting Groups, S. 38–39.
- ↑ a b P.J. Kocieński: Protecting Groups, S. 43.
- ↑ J.F.W. McOmie, D.E. West: „3,3'-Dihyroxybiphenyl“, in: Org. Synth., 1969, 49, S. 50.
- ↑ P.J. Kocieński: Protecting Groups, S. 46–49.
- ↑ Yuji Oikawa, Tadao Yoshioka, Osamu Yonemitsu: „Specific removal of o-methoxybenzyl protection by DDQ oxidation“, in: Tetrahedron Lett., 1982, 23, S. 885–888; doi:10.1016/S0040-4039(00)86974-9.
- ↑ Rolf Johansson, Bertil Samuelsson: „Regioselective reductive ring-opening of 4-methoxybenzylidene acetals of hexopyranosides. Access to a novel protecting-group strategy. Part 1“, in: J. Chem. Soc., Perkin Trans. 1, 1984, S. 2371–2374; doi:10.1039/P19840002371.
- ↑ Literatur wie p-Methoxybenzyl.
- ↑ Michel Bessodes, Dimitri Komiotis, Kostas Antonakis: „Rapid and selective detritylation of primary alcohols using formic acid“, in: Tetrahedron Lett., 1986, 27, S. 579–580; doi:10.1016/S0040-4039(00)84045-9.
- ↑ B. Helferich: Carbonhydr. Chem. Biochem., 1948, 3, S. 79.
- ↑ M.L. García, J. Pascual, L. Borràs, J.A. Andreu, E. Fos, D. Mauleón, G. Carganico, F. Arcamone: „Synthesis of new ether glycerophospholipids structurally related to modulator“, in: Tetrahedron, 1991, 47, S. 10023–10034; doi:10.1016/S0040-4020(01)96051-X.
- ↑ P.J. Kocieński: Protecting Groups, S. 59–60.
- ↑ P.J. Kocieński: Protecting Groups, S. 62.
- ↑ R.E. Ireland, D.W. Norbeck: „Convergent synthesis of polyether ionophore antibiotics: the synthesis of the monensin bis(tetrahydrofuran) via the Claisen rearrangement of an ester enolate with a β-leaving group“, in: J. Am. Chem. Soc., 1985, 107, S. 3279–3285; doi:10.1021/ja00297a038.
- ↑ Literatur siehe Allyl.
- ↑ Paul A. Wender, Carlos R. D. Correia: „Intramolecular photoinduced diene-diene cyaloadditions: a selective method for the synthesis of complex eight-membered rings and polyquinanes“, in: J. Am. Chem. Soc., 1987, 109, S. 2523–2525; doi:10.1021/ja00242a053.
- ↑ Elias J. Corey, Mark G. Bock: „Protection of primary hydroxyl groups as methylthiomethyl ethers“, in: Tetrahedron Lett., 1975, 16, S. 3269–3270; doi:10.1016/S0040-4039(00)91422-9.
- ↑ Elias J. Corey, Duy H. Hua, Bai Chuan Pan, Steven P. Seitz: „Total synthesis of aplasmomycin“, in: J. Am. Chem. Soc., 1982, 104, S. 6818–6820; doi:10.1021/ja00388a074.
- ↑ Serge David, Annie Thieffry, Alain Veyrières: „A mild procedure for the regiospecific benzylation and allylation of polyhydroxy-compounds via their stannylene derivatives in non-polar solvents“, in: J. Chem. Soc., Perkin Trans. 1, 1981, S. 1796–1801; doi:10.1039/P19810001796.
- ↑ Kaoru Fuji, Shigetoshi Nakano, Eiichi Fujita: „An Improved Method for Methoxymethylation of Alcohols under Mild Acidic Conditions“, in: Synthesis, 1975, S. 276–277.
- ↑ P.J. Kocieński: Protecting Groups, S. 77.
- ↑ H. Nagaoka, W. Rutsch, G. Schmidt, H. Ito, M.R. Johnson, Y. Kishi: „Total synthesis of rifamycins. 1. Stereocontrolled synthesis of the aliphatic building block“, in: J. Am. Chem. Soc., 1980, 102, S. 7962–7965; doi:10.1021/ja00547a037.
- ↑ W. Clark Still, Dominick Mobilio: „Synthesis of asperdiol“, in: J. Org. Chem., 1983, 48, S. 4785–4786; doi:10.1021/jo00172a070.
- ↑ Masahiro Hirama, Mitsuko Uei: „A chiral total synthesis of compactin“, in: J. Am. Chem. Soc., 1982, 104, S. 4251–4253; doi:10.1021/ja00379a037.
- ↑ W. Clark Still, Shizuaki Murata, Gilbert Revial, Kazuo Yoshihara: „Synthesis of the cytotoxic germacranolide eucannabinolide“, in: J. Am. Chem. Soc., 1983, 105, S. 625–627; doi:10.1021/ja00341a055.
- ↑ Robert C. Gadwood, Renee M. Lett, Jane E. Wissinger: „Total synthesis of (±)-poitediol and (±)4-epipoitediol“, in: J. Am. Chem. Soc., 1984, 106, S. 3869–3870; doi:10.1021/ja00325a032.
- ↑ Steven D. Burke, Gregory J. Pacofsky: „The ester enolate claisen rearrangement. Total synthesis of (±)-ethisolide“, in: Tetrahedron Lett., 1986, 27, S. 445–448; doi:10.1016/S0040-4039(00)85501-X.
- ↑ Toshiyuki Kan, Masaru Hashimoto, Mitsutoshi Yanagiya, Haruhisa Shirahama: „Effective deprotection of 2-(trimethylsilylethoxy)methylated alcohols (SEM ethers). Synthesis of thyrsiferyl-23 acetate“, in: Tetrahedron Lett., 1988, 29, S. 5417–5418; doi:10.1016/S0040-4039(00)82883-X.
- ↑ Joseph P. Marino, Scott L. Dax: „An efficient desilylation method for the generation of o-quinone methides: application to the synthesis of (+)- and (−)-hexahydrocannabinol“, in: J. Org. Chem., 1984, 49, S. 3671–3672; doi:10.1021/jo00193a051.
- ↑ Karel F. Bernady, M. Brawner Floyd, John F. Poletto, Martin J. Weiss: „Prostaglandins and congeners. 20. Synthesis of prostaglandins via conjugate addition of lithium trans-1-alkenyltrialkylalanate reagents. A novel reagent for conjugate 1,4-additions“, in: J. Org. Chem., 1979, 44, S. 1438–1447; doi:10.1021/jo01323a017.
- ↑ Elias J. Corey, Haruki Niwa, Jochen Knolle: „Total synthesis of (S)-12-hydroxy-5,8,14-cis,-10-trans-eicosatetraenoic acid (Samuelsson's HETE)“, in: J. Am. Chem. Soc., 1978, 100, S. 1942–1943; doi:10.1021/ja00474a058.
- ↑ a b P. Collins, R. Ferrier: Monosacharides – Their Chemistry and their Roles in Natural Products, Wiley, West Sussex 1995, ISBN 0-471-95343-1.
- ↑ Christopher R. Schmid, Jerry D. Bryant: D-(R)-Glycerinaldehyd Acetonide, in: Org. Synth., 1995, 72, S. 6.
- ↑ András Lipták, János Imre, János Harangi, Pál Nánási, András Neszmélyi: „Chemo-, stereo- and regioselective hydrogenolysis of carbohydrate benzylidene acetals. Synthesis of benzyl ethers of benzyl α-D-, methyl β-D-mannopyranosides and benzyl α-D-rhamnopyranoside by ring cleavage of benzylidene derivatives with the LiAlH4-AlCl3 reagent“, in: Tetrahedron, 1982, 38, S. 3721–3727; doi:10.1016/0040-4020(82)80083-5.
- ↑ James A. Marshall, Joseph D. Trometer, Bruce E. Blough, Thomas D. Crute: "Stereochemistry of SN2' additions to acyclic vinyloxiranes", in J. Org. Chem. 1988, 53, 4274–4282 doi:10.1021/jo00253a020
- ↑ T. Tsunoda, M. Suzuki, R. Noyori: „A facile procedure for acetalization under aprotic conditions“, in: Tetrahedron Lett., 1980, 21, S. 1357–1358; doi:10.1016/S0040-4039(00)74575-8.
- ↑ Juji Yoshimura, Shigeomi Horito, Hiroriobu Hashimoto: „Facile Synthesis of 2,3,4,6-Tetra-O-benzyl-D-glucopyranosylidene Acetals Using Trimethylsilyl Trifluoromethanesulfonate Catalyst“, in: Chem. Lett., 1981, 10, S. 375–376; doi:10.1246/cl.1981.375.
- ↑ Bruce H. Lipshutz, Daniel Pollart, Joseph Monforte, Hiyoshizo Kotsuki: „Pd(II)-catalyzed acetal/ketal hydrolysis/exchange reactions“, in: Tetrahedron Lett., 1985, 26, S. 705–708; doi:10.1016/S0040-4039(00)89114-5.
- ↑ Kwan Soo Kim, Yang Heon Song, Bong Ho Lee, Chi Sun Hahn: „Efficient and selective cleavage of acetals and ketals using ferric chloride adsorbed on silica gel“, in: J. Org. Chem., 1986, 51, S. 404–407; doi:10.1021/jo00353a027.
- ↑ P.J. Kocieński: Protecting Groups, S. 167–170.
- ↑ P.J. Kocieński: Protecting Groups, S. 164–167.
- ↑ P.J. Kocieński: Protecting Groups, S. 176.
- ↑ P.J. Kocieński: Protecting Groups, S. 178–180.
- ↑ Samuel J. Danishefsky, Nathan B. Mantlo, Dennis S. Yamashita, Gayle. Schulte: „Concise route to the calichemicin-esperamicin series: the crystal structure of an aglycone prototype“, in: J. Am. Chem. Soc., 1988, 110, S. 6890–6891; doi:10.1021/ja00228a051.
- ↑ John N. Haseltine, Maria Paz Cabal, Nathan B. Mantlo, Nobuharu Iwasawa, Dennis S. Yamashita, Robert S. Coleman, Samuel J. Danishefsky, Gayle K. Schulte: „Total synthesis of calicheamicinone: new arrangements for actuation of the reductive cycloaromatization of aglycon congeners“, in: J. Am. Chem. Soc. Nr. 113, 1991, S. 3850–3866; doi:10.1021/ja00010a030.
- ↑ P.J. Kocieński: Protecting Groups, S. 119.
- ↑ Satomi Niwayama: „Highly Efficient Selective Monohydrolysis of Symmetric Diesters“, in: J. Org. Chem., 2000, 65, S. 5834–5836; doi:10.1021/jo0001986.
- ↑ J.M. Khurana, Arti Sehgal: „An efficent and convenient procedure for ester hydrolysis“, in: Org. Prep. Proced. Ind., 1994, 26, S. 580–583.
- ↑ Robert V. Stevens, Albert W. M. Lee: „Stereochemistry of the Robinson-Schoepf reaction. A stereospecific total synthesis of the ladybug defense alkaloids precoccinelline and coccinelline“, in: J. Am. Chem. Soc., 1979, 101, S. 7032–7035; doi:10.1021/ja00517a042.
- ↑ J. Wrobel, K. Takahashi, V. Honkan, G. Lannoye, J. M. Cook, Steven H. Bertz: „α-Lithio ketones. 1. Stereocontrolled synthesis of (±)-modhephene via the Weiss reaction“, in: J. Org. Chem., 1983, 48, S. 139–141; doi:10.1021/jo00149a034.
- ↑ Dennis D. Keith, John A. Tortora, Roxana Yang: „Synthesis of L-2-amino-4-methoxy-trans-but-3-enoic acid“, in: J. Org. Chem., 1978, 43, S. 3711–3713; doi:10.1021/jo00413a016.
- ↑ Peter Mohr, Nada Waespe-Šarčević, Christoph Tamm, Krystyna Gawronska, Jacek K. Gawronski: „A Study of Stereoselective Hydrolysis of Symmetrical Diesters with Pig Liver Esterase“, in: Helv. Chim. Acta, 1983, 66, S. 2501–2511; doi:10.1002/hlca.19830660815.
- ↑ Théophile Tschamber, Nada Waespe-Šarčević, Christoph Tamm: „Stereocontrolled Synthesis of an Epimer of the C(19)-to-C(27) Segment of Rifamycin S“, in: Helv. Chim. Acta, 1986, 69, S. 621–625; doi:10.1002/hlca.19860690311.
- ↑ Yves Rubin, Carolyn B. Knobler, Francois Diederich: „Precursors to the cyclo[n]carbons: from 3,4-dialkynyl-3-cyclobutene-1,2-diones and 3,4-dialkynyl-3-cyclobutene-1,2-diols to cyclobutenodehydroannulenes and higher oxides of carbon“, in: J. Am. Chem. Soc., 1990, 112, S. 1607–1617; doi:10.1021/ja00160a047.
- ↑ Sunggak Kim, Yong Gil Kim, Deog-il Kim: „A novel method for selective dioxolanation of ketones in the presence of aldehydes“, in: Tetrahedron Lett., 1992, 33, S. 2565–2566; doi:10.1016/S0040-4039(00)92243-3.
- ↑ G. Bauduin, D. Bondon, Y. Pietrasanta, B. Pucci: „Reactions de transcetalisation – II: Influence des facteurs steriques et electroniques sur les energies de cetalisation“, in: Tetrahedron, 1978, 34, S. 3269–3274; doi:10.1016/0040-4020(78)80243-9.
- ↑ John E. McMurry, Stephen J. Isser: „Total synthesis of longifolene“, in: J. Am. Chem. Soc., 1972, 94, S. 7132–7137; doi:10.1021/ja00775a044.
- ↑ M.P. Bosch, M. Pilar Bosch, Francisco Camps, Jose Coll, Angel Guerrero, Toshio Tatsuoka, Jerrold Meinwald: „A stereoselective total synthesis of (±)-muzigadial“, in: J. Org. Chem., 1986, 51, S. 773–784; doi:10.1021/jo00356a002.
- ↑ Ulrich Schmidt, Thomas Beuttler, Albrecht Lieberknecht, Helmut Griesser: „Aminosäuren und peptide – XXXXII. Synthese von Chlamydocin + epi-Chlamydocin“, in: Tetrahedron Lett., 1983, 24, S. 3573–3576; doi:10.1016/S0040-4039(00)88171-X.
- ↑ Elias J. Corey, Plato A. Magriotis: „Total synthesis and absolute configuration of 7,20-diisocyanoadociane“, in: J. Am. Chem. Soc., 1987, 109, S. 287–289; doi:10.1021/ja00235a052.
- ↑ Elias J. Corey, Kyriacos C. Nicolaou, Takeshi Toru: „Total synthesis of (±)-vermiculine“, in: J. Am. Chem. Soc., 1975, 97, S. 2287–2288; doi:10.1021/ja00841a058.
- ↑ Tainejiro Hiyama, Akihiro Kanakura, Hajime Yamamoto, Hitosi Nozaki: „General Route to α,β-unsaturated Aldehydes of Homoterpenoid and terpenoid Structure. Sythesis of JH-II and β-Sinensal“, in: Tetrahedron Lett., 1978, 19, S. 3051–3054; doi:10.1016/S0040-4039(01)94936-6.
- ↑ F. Huet, A. Lechevallier, M. Pellet, J.M. Conia: „Wet Silica Gel; A Convenient Reagent for Deacetalization“, in: Synthesis, 1978, S. 63–64.
- ↑ F. Zymalkokowski: Katalytische Hydrierung, Ferdinand Enke Verlag, Stuttgart 1965, S. 127–133.
- ↑ P.J. Kocieński: Protecting Groups, S. 136.
- ↑ P.J. Kocieński: Protecting Groups, S. 139–142.
- ↑ Ahmed M. Tafesh, Jens Weiguny: „A Review of the Selective Catalytic Reduction of Aromatic Nitro Compounds into Aromatic Amines, Isocyanates, Carbamates, and Ureas Using CO“, in: Chem. Rev., 1996, 96, S. 2035–2052; doi:10.1021/cr950083f.
- ↑ Evan L. Allred, Boyd R. Beck, Kent J. Voorhees: „Formation of carbon-carbon double bonds by the reaction of vicinal dihalides with sodium in ammonia“, in: J. Org. Chem., 1974, 39, S. 1426–1427; doi:10.1021/jo00926a024.
- ↑ Timothy S. Butcher, Feng Zhou, Michael R. Detty: „Debrominations of vic-Dibromides with Diorganotellurides. 1. Stereoselectivity, Relative Rates, and Mechanistic Implications“, in: J. Org. Chem., 1998, 63, S. 169–176; doi:10.1021/jo9713363.
- ↑ C. J. Li, David N. Harpp: „Bis(triphenylstanyl)telluride a mild and selective reagent for telluration and debromination“, in: Tetrahedron Lett., 1990, 31, S. 6291–6293; doi:10.1016/S0040-4039(00)97045-X.
- ↑ Corrado Malanga, Serena Mannucci, Luciano Lardicci: „Carbon-halogen bond activation by nickel catalyst: Synthesis of alkenes, from 1,2-dihalides“, in: Tetrahedron, 1998, 54, S. 1021–1028; doi:10.1016/S0040-4020(97)10203-4.
- ↑ Byung Woo Yoo, Seo Hee Kim, Jun Ho Kim: „A Mild, Efficient, and Selective Debromination of vic-Dibromides to Alkenes with Cp2TiCl2/Ga System“, in: Bull. Korean Chem. Soc., 2010, 31, S. 2757–2758; doi:10.5012/bkcs.2010.31.10.2757.
- ↑ Antonius J. H. Klunder, Jie Zhu, Binne Zwanenburg: „The Concept of Transient Chirality in the Stereoselective Synthesis of Functionalized Cycloalkenes Applying the Retro-Diels−Alder Methodology“, in: Chem. Rev., 1999, 99, S. 1163–1190; doi:10.1021/cr9803840.
- ↑ Hideyuki Tanaka, Takashi Kamikubo, Naoyuki Yoshida, Hideki Sakagami, Takahiko Taniguchi, Kunio Ogasawara: „Enantio- and Diastereocontrolled Synthesis of (−)-Iridolactone and (+)-Pedicularis-lactone“, in: Org. Lett., 2001, 3, S. 679–681; doi:10.1021/ol0070029.
- ↑ Martin Banwell, David Hockless, Bevyn Jarrott, Brian Kelly, Andrew Knill, Robert Longmore, Gregory Simpson: „Chemoenzymatic approaches to the decahydro-as-indacene cores associated with the spinosyn class of insecticide“, in: J. Chem. Soc., Perkin Trans. 1, 2000, S. 3555–3558; doi:10.1039/b006759h.
- ↑ Wenzel E. Davidsohn, Malcolm C. Henry: „Organometallic Acetylenes of the Main Groups III–V“, in: Chem. Rev., 1967, 67, S. 73–106; doi:10.1021/cr60245a003.
- ↑ Barry J. Teobald: „The Nicholas reaction: the use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis“, in: Tetrahedron, 2002, 58, S. 4133–4170; doi:10.1016/S0040-4020(02)00315-0.
- ↑ Kenneth M. Nicholas, R. Pettit: „An alkyne protection group“, in: Tetrahedron Lett., 1971, 37, S. 3475–3478; doi:10.1016/S0040-4039(01)97209-0.
- ↑ Richard E. Connor, Kenneth M. Nicholas: „Isolation, characterization, and stability of α-[(ethynyl)dicobalt hexacarbonyl] carbonium ions“, in: J. Organomet. Chem., 1977, 125, C45–C48; doi:10.1016/S0022-328X(00)89454-1.
- ↑ Rosa F. Lockwood, Kenneth M. Nicholas: „Transition metal-stabilized carbenium ions as synthetic intermediates. I. α-[(alkynyl)dicobalt hexacarbonyl] carbenium ions as propargylating agents“, in: Tetrahedron Lett., 1977, S. 4163–4166; doi:10.1016/S0040-4039(01)83455-9.
- ↑ K.M. Nicholas, R. Pettit: „On the stability of α-(alkynyl)dicobalt hexacarbonyl carbonium ions“, in: J. Organomet. Chem., 1972, 44, C21–C24; doi:10.1016/0022-328X(72)80037-8.
- ↑ T. Reichstein, A. Grüssner: „Eine ergiebige Synthese der L-Ascorbinsäure (C-Vitamin)“, in: Helv. Chim. Acta, 1934, 17, S. 311–328; doi:10.1002/hlca.19340170136.
- ↑ K.C. Nicolaou, E.J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods, VCH Verlagsgesellschaft mbH, Weinheim 1996, S. 711–729, ISBN 3-527-29284-5.
- ↑ Peter G.M. Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis, 4th Ed., John Wiley & Sons Inc., Hoboken, New Jersey, S. 10–13; ISBN 0-471-69754-0.
- ↑ J.M. McClure, Samuel J. Danishefsky: „A novel Heck arylation reaction: rapid access to congeners of FR 900482“, in: J. Am. Chem. Soc., 1993, 115, S. 6094–6100; doi:10.1021/ja00067a026.
- ↑ Serge L. Beaucage, Radhakrishman P. Iyer: „Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach“, in: Tetrahedron, 1992, 48, S. 2223–2311; doi:10.1016/S0040-4020(01)88752-4.
- ↑ Michael Schelhaas, Herbert Waldmann: „Schutzgruppenstrategien in der organischen Synthese“, in: Angewandte Chemie, 1996, 103, S. 2194; doi:10.1002/ange.19961081805.
- ↑ Marco Eissen, Radoslaw Mazur, Heinz-Georg Quebbemann und Karl-Heinz Pennemann: „Atom Economy and Yield of Synthesis Sequences“, in: Helv. Chim. Acta, 2004, 87, S. 524–535; doi:10.1002/hlca.200490050.
- ↑ Marco Eissen, Jürgen O. Metzger: „Environmental Performance Metrics for Daily Use in Synthetic Chemistry“, in: Chemistry a European Journal, 2002, 8, S. 3580–3585; doi:10.1002/chin.200247273.
- ↑ Marco Eissen, Jürgen O. Metzger, Eberhard Schmidt, Uwe Schneidewind: 10 Jahre nach „Rio“ – Konzepte zum Beitrag der Chemie zu einer nachhaltigen Entwicklung, in: Angewandte Chemie, 2002, 114, S. 402–425; doi:10.1002/1521-3757(20020201)114:3<402::AID-ANGE402>3.0.CO;2-D.
- ↑ Siegfried Hauptmann: Organische Chemie, 2. Auflage, VEB Deutscher Verlag für Grundstoffindustrie, Leipzig 1985, ISBN 3-342-00280-8, S. 621–622.
- ↑ Hideaki Muratake, Harumi Kumagami, Mitsutaka Natsume: Synthetic studies of marine alkaloids hapalindoles. Part 3 Total synthesis of (±)-hapalindoles H and U in Tetrahedron 1990, 46, 6351-6360 doi:10.1016/S0040-4020(01)96007-7.
- ↑ Hideaki Muratake, Mitsutaka Natsume: Synthetic studies of marine alkaloids hapalindoles. Part I Total synthesis of (±)-hapalindoles J and M in Tetrahedron 1990, 46, 6331-6342 doi:10.1016/S0040-4020(01)96005-3.
- ↑ Hideaki Muratake, Mitsutaka Natsume: Synthetic studies of marine alkaloids hapalindoles. Part 2. Lithium aluminum hydride reduction of the electron-rich carbon-carbon double bond conjugated with the indole nucleus in Tetrahedron 1990, 46, 6343-6350 doi:10.1016/S0040-4020(01)96006-5.
- ↑ Phil S. Baran, Thomas J. Maimone, Jeremy M. Richter: Total synthesis of marine natural products without using protecting groups in Nature 446, 404-408 doi:10.1038/nature05569.
- ↑ B. Speiser: Elektrodenreaktionen und Chronoamperometrie, Chemie in unserer Zeit, 15. Jahrg. 1981, Nr. 1, S.21