Polymeranaloge Reaktion
Eine polymeranaloge Reaktion ist eine Reaktion, bei der an Polymeren eine funktionelle Gruppe FG1 durch eine chemische Reaktion in eine andere funktionelle Gruppe FG2 überführt wird:[1]

Dabei werden grundsätzlich zwei Arten von polymeranalogen Reaktionen unterschieden:
- Polymeranaloge Reaktionen im klassischen Sinn (Polymertransformationen), bei denen das Reaktionsprodukt das gewünschte Polymer ist
- Reaktionen an Reaktivpolymeren. Dies sind meist vernetzte Polymere mit funktionellen Gruppen, mit denen andere, meist niedermolekulare Verbindungen hergestellt werden können. Das Reaktivpolymer kann anschließend meist wieder regeneriert werden. Das bekannteste Beispiel sind Ionenaustauscher, bei denen meist niedermolekulare Ionen ausgetauscht werden.
Bei diesen Reaktionen ändert sich die molare Masse und gegebenenfalls auch die Konstitution der Polymere, der Polymerisationsgrad bleibt aber erhalten.[1] Eine komplette Umsetzung der reaktiven Gruppen ist normalerweise nicht möglich, eine Ausnahme bilden Reaktivpolymere und Ionenaustauscher, die durch die Durchführung der Umsetzung sehr hohe Umsätze ermöglichen. In vielen Fällen ist eine komplette Umsetzung zudem nicht gewünscht, hier bilden Polyvinylalkohol/Polyvinylamin Ausnahmen, bei denen man neben teilhydrolysierten auch möglichst komplett hydrolysierte Typen anstrebt. Da sich die physikalischen und chemischen Eigenschaften der Produkte mit dem Substitutionsgrad ändern, versucht man in Fällen wo bestimmte Substitutionsgrade angestrebt werden diese gezielt durch die Reaktionsführung zu erreichen, um die gewünschten Eigenschaften zu erhalten. So sinkt in der Regel ab einem bestimmten Substitutionsgrad bei Cellulose- und Stärkederivaten die Löslichkeit bzw. Quellfähigkeit und auch die biologische Abbaubarkeit. Ab welchem Grad dies geschieht, hängt u. a. von der Größe und Hydrophobie des Substituenten ab.
Von der polymeranalogen Reaktion ist die Vernetzung zu unterscheiden. Hier reagiert ein Polymer mit einem niedermolekularen Vernetzer oder einem anderen Polymer zu größeren Aggregaten, die nach der Reaktion eine weit größere Molmasse und Polymerisationsgrad haben als das Ausgangspolymer.
Geschichte Bearbeiten
Bis ins 19. Jahrhundert hinein wurden natürliche Polymere wie Baumwolle, Wolle, Seide und Leinen selten gezielt chemisch behandelt, um ihre Eigenschaften zu verändern. Nur beim Färben wurde, abhängig von der Faser und dem Farbstoff bzw. der Färbemethode, mit Laugen, Salzen oder anderen Substanzen ein besseres Färbeverhalten erzielt, das sog. Beizen.[2] Ab Mitte des 19. Jahrhunderts wurden Cellulosederivate wie 1846 die Schießbaumwolle (Christian Friedrich Schönbein[3]) und 1865 das Celluloseacetat (Paul Schützenberger[4]) gewonnen. Erst im 20. Jahrhundert, als von Hermann Staudinger die Natur der Polymere aufgeklärt wurde und erste künstliche Polymere wie Bakelit (1909), Polyvinylchlorid (ab 1913), Polyester (ab den 1920ern), Polyethylen (ab 1933) und Polyamide (ab 1935) in größeren Mengen hergestellt und verwendet wurden, kamen Methoden auf, diese Polymere durch gezielte chemische Behandlung zu modifizieren.
Wirtschaftliche Bedeutung Bearbeiten
Da es eine Vielzahl von Anwendungen gibt, die in einem breiten Feld von Reaktionsarten und Anwendungsgebieten liegen, lässt es sich nur schwer quantitativ abschätzen, welche Umsatzzahlen Prozesse, die polymeranaloge Reaktionen beinhalten haben, zumal eine Polymeranaloge Reaktion oft nur einen Teil der Wertschöpfungskette darstellt. Die DECHEMA hat im Jahr 2004 in einem Positionspapier die Polymer-Modifikation in Extrudern/Schneckenmaschinen als Spezialfall der Polymeranalogen Reaktion als besonders vielversprechende Technologie herausgestellt.[5][6] Aus der Vielzahl der Anwendungen lässt sich aber grob abschätzen, dass es sich jährlich weltweit um dreistellige Millionen bis Milliardenbeträgen (in €) handelt.
Reaktionskinetik bei polymeranalogen Reaktionen Bearbeiten
Im Gegensatz zu den meist homogenen Reaktionen in Lösemitteln liegen bei polymeranalogen Reaktionen meist heterogene Verhältnisse vor. Im Inneren des Polymerknäuels ist die Konzentration reaktiver, polymergebundener Gruppen hoch, im umgebenden Lösemittel sehr niedrig, bis zu Null. Zudem kann es durch die räumliche Nachbarschaft von noch nicht reagierten Gruppen zu schon reagierten zu Nachbargruppeneffekten kommen, die reaktionsbeschleunigend, oder reaktionsverlangsamend wirken können.[7] Folgende Fälle können unterschieden werden:
- Elektronische Effekte[7]
- Sterische Effekte
- Löslichkeitseffekte
Künstliche Polymere Bearbeiten
Polymere nicht existenzfähiger Monomere Bearbeiten
Durch polymeranaloge Reaktion werden Polymere hergestellt, die nicht direkt aus den (formalen) Monomeren synthetisiert werden können, weil diese Monomere nicht stabil oder existent sind, oder die formalen Monomere andere Polymere liefern als die gewünschten.
Polyvinylalkohol Bearbeiten
Ein kommerziell wichtiges Beispiel ist Polyvinylalkohol (PVA). Der hypothetisch zugrunde liegende Vinylalkohol liegt in einem tautomeren Gleichgewicht mit Acetaldehyd vor, wobei die Gleichgewichtlage nahezu vollständig auf Seiten des Aldehyds liegt:[8]

PVA wird hergestellt, indem aus dem stabilen Monomer Vinylacetat zuerst Polyvinylacetat hergestellt wird. Aus diesem wird mit Butanol oder Methanol durch eine Umesterung der Polyvinylalkohol erhalten. Die dabei anfallenden Ester (Butylacetat und Methylacetat) sind wertvolle Lösemittel.[9] Meist wird eine möglichst quantitative Umesterung angestrebt, Es gibt aber auch teilweise hydrolysierte Polyvinylalkohole, die beispielsweise als Klebstoffe Verwendung finden.[10] Die Löslichkeit in Wasser hängt aber neben dem Hydrolysegrad von anderen Faktoren wie Molmasse und Taktizität ab:[11]

Polyvinylamin Bearbeiten
Ähnliches gilt für Polyvinylamin, das Vinylamin läge auch hier mit Ethylidenimin in einem Gleichgewicht vor, in diesem Fall einem Imin-Enamin-Tautomerie-Gleichgewicht, allerdings sind beide Verbindungen instabil.[12]

Polyvinylamin wird aus N-Vinylformamid hergestellt, das zu Polyvinylformamid polymerisiert und durch dessen Verseifung gewonnen wird.

Polyethylenimin Bearbeiten
Mit p-Toluolsulfonsäuremethylester als Initiator lassen sich 2-alkyl-substituierte 2-Oxazoline zu N-substituierten Polyethylenimin polymerisieren. Nach Verseifung entsteht daraus ein lineares Polyethylenimin.[13]

Nachbehandlung von Polymeren Bearbeiten
- Polyethylen, Ethylen-Propylen-Copolymere, Polyvinylchlorid und andere Polymere werden nach ihrer Herstellung chloriert, um mechanische und chemische Eigenschaften zu verbessern. Zur Verbesserung der Elastomereigenschaften sollte der Chlorgehalt des Polymers 25–40 % betragen. Soll das Polymer zur Verbesserung der Schlagfestigkeit mit PVC geblendet werden, sollten es > 40 % sein. Hochchlorierte PVC-Typen mit bis zu 65 % Chlor werden in Lacken und Klebstoffen verwendet, wobei deren Verwendung deutlich rückläufig ist, bzw. sich auf Spezialanwendungen reduziert.[14]
Nachbehandlung von Polyethylen Bearbeiten
Polyethylen wird in Suspension – katalysiert durch Schwermetallsalze – chloriert. Dabei strebt man meist einen Substitutionsgrad von < 30 % an.[15]

Nachbehandlung von Ethylen-Propylen-Copolymeren Bearbeiten

Nachbehandlung von Polyvinylchlorid (PVC) Bearbeiten
PVC wird in Lösung bis zu einem Chlorgehalt von ca. 64 % chloriert:[15]

Das Reaktionsprodukt wird für die Herstellung von Fasern, Lacken und Klebstoffen genutzt.[15]
Nachbehandlung von Acrylnitril-Butadien-Kautschuk Bearbeiten
Acrylnitril-Butadien-Kautschuk wird zur Verbesserung der Alterungsbeständigkeit hydriert:[14]

Ionenaustauscher Bearbeiten
Ionenaustauscher sind meist vernetzte Polystyrolharze oder Cellulose, die anionische oder kationische Gruppen tragen. Als anionische Gruppe dient bei den starken Kationenaustauschern meist eine Sulfonsäuregruppe, bei den schwachen eine Carboxylatgruppe. Die kationischen Gruppen sind je nach Anwendung stark basische quartäre Ammoniumverbindungen, oder tertiäre, sekundäre oder primäre (= schwach basische) Amine.[16]
Starke Kationenaustauscher Bearbeiten
-
 Saure Form der Sulfonsäure
Saure Form der Sulfonsäure -
 Neutrale Form der Sulfonsäure
Neutrale Form der Sulfonsäure -
 Basische Form der Ammoniumverbindung
Basische Form der Ammoniumverbindung -
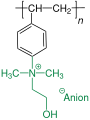 Neutrale Form der Ammoniumverbindung
Neutrale Form der Ammoniumverbindung




Schwache Ionenaustauscher Bearbeiten
Bei der polymeranalogen Reaktion von Cellulose werden nicht alle Wasserstoffatome ersetzt, sondern es entstehen mehrere Reaktionsprodukte. Diese enthalten wie in den schemenhaften Abbildungen gezeigt unterschiedlich viele Ammoniumverbindungen und Carboxylatgruppen.
Schematische Darstellung für einen Anionenaustauscher:

Schematische Darstellung für einen Kationenaustauscher:

Reaktivpolymere Bearbeiten
- Neben Ionenaustauschern gibt es eine ganze Reihe (vernetzter) Polymere, die funktionelle Gruppen tragen, mit denen niedermolekulare Verbindungen hergestellt, oder umgesetzt werden können. Basis bilden meist vernetzte Polystyrole. Beispiele sind:[17]

Die Reaktionen an den Reaktivpolymeren und deren Regeneration erfolgen analog wie bei Ionenaustauschern in Säulen, was durch die großen Konzentrationsgradienten sowohl die Reaktion als auch die Regeneration mit großen Ausbeuten durchführbar machen.
- Die Reaktionen verlaufen bei polymer gebundenen Gruppen nicht immer analog zu den Reaktionen monomerer reaktiver Gruppen. So bromiert N-Bromsuccinimid Olefine in der Allylstellung unter Erhalt der Doppelbindung, während das polymer gebundene zu einer Addition von Brom an die Doppelbindung führt.[17]

Merrifield-Synthese Bearbeiten
Bei der Merrifield-Synthese wird an einem vernetzten Polystyrol mit einer Chlormethyl-Gruppe (CH2-Cl) schrittweise ein Peptid synthetisiert.[18]

Die Sequenz beginnt damit, dass eine am N-Terminus geschützte Aminosäure an die CH2-Cl Gruppe des Harzes gekoppelt und anschließend die Schutzgruppe entfernt wird. An diese Aminogruppe kann wieder eine Aminosäure unter Ausbildung einer Peptidbindung gekuppelt werde. Um den Umsatz hoch zu gestalten, wird üblicherweise mit einem großen Überschuss der zu koppelnden Aminosäure gearbeitet. Durch Wiederholung diese Sequenz können Peptide mit einer Länge von maximal ca. 100 Aminosäuren hergestellt werden.[19]

Seitdem Peptide routinemäßig mit genetischen Methoden hergestellt werden können, hat die Merrifield-Synthese, von Spezialfällen wie beispielsweise dem Einbau von nichtkanonischen Aminosäuren abgesehen, keine praktische Bedeutung mehr.
Leiterpolymere Bearbeiten
Durch eine intramolekulare Polymerisation kann man geeignete Polymere zu Leiterpolymeren umsetzen. Ein geeignetes Grundpolymer ist beispielsweise isotaktisches 1,2-Polybutadien, bei dem die seitenständigen Vinylgruppen cyclisiert werden.[20] Die folgende Abbildung gibt einen schematisierten und idealisierten Ablauf der Reaktion an.

Natürliche Polymere Bearbeiten
Cellulosederivate Bearbeiten
Polymeranaloge Reaktionen bei nativer, oder manchmal auch gezielt abgebauter Cellulose liefern wichtige Produkte der Kunststoffindustrie.
Bei technischen Produkten liegt der Substitutionsgrad meist zwischen zwei und fünf pro Cellobioseeinheit und wird gezielt angestrebt, weil die unterschiedlichen Substitutionsgrade den Derivaten unterschiedliche Eigenschaften verleihen.
Celluloseester Bearbeiten
- Celluloseacetat, einer der ältesten Kunststoffe, wird je nach Substitutionsgrad nach unterschiedlichen Verfahren hergestellt, ausführlich werden sie im Hauptartikel dazu beschrieben. Da sich Fasern aus Celluloseacetat ähnlich anfühlen wie Seide und auch im Aussehen ähnlich sind, wird sie in großem Umfang zur Herstellung dieser Fasern und Bekleidung daraus verwendet, zumal diese Stoffe pflegeleichter und unempfindlicher sind als Seide.[21]

- Cellulosenitrat gehört auch zu den ältesten Kunststoffen. Mit Campher plastifiziert wurde es zur Herstellung von Zelluloid verwendet, einem der ersten Thermoplaste. Es wird heute allerdings wegen der großen Brandgefahr nur noch selten verwendet, beispielsweise für die Herstellung von Tischtennisbällen[22][23] und von Sprengstoff. Cellulosenitrat unterliegt dem deutschen Sprengstoffgesetz. Bei einem Stickstoffgehalt > 12,75 % handelt es sich dann überwiegend um Cellulosetrinitrat (Schießbaumwolle), bei einem Gehalt < 12,75 % um Cellulosedinitrat (Kollodiumwolle).

Celluloseether Bearbeiten
- Hydroxypropylcellulose[24] wird aus alkalisch vorbehandelter Cellulose und Propylenoxid hergestellt. Sie wird als Emulgator, Verdickungsmittel und Bindemittel verwendet.

Da bei dieser Reaktion nicht alle Hydroxygruppen reagieren, entstehen Gemische mit unterschiedlich hohem Substitutionsgrad. Auch der Substitutionsgrad der einzelnen Stärkebausteine innerhalb eines Polymers kann unterschiedlich hoch ausfallen. Analoge Gemische entstehen bei den folgenden Reaktionen.
- Methylcellulose wird durch Umsetzen von Methylchlorid mit alkalisch vorbehandelter Cellulose hergestellt und als Verdickungsmittel, speziell als Tapetenkleister verwendet.[25]
- Ethylcellulose wird durch Umsetzen von Ethylchlorid mit alkalisch vorbehandelter Cellulose hergestellt. Sie ist beispielsweise ein Bestandteil von Celluloseetherlacken.[26]
- Hydroxypropylmethylcellulose stellt man aus einer alkalisch vorbehandelten Cellulose mit einer Mischung aus Propylenoxid und Methylchlorid her. Die Verwendung ist vielfältig. Unter anderem wird sie als Verdickungsmittel[27] eingesetzt.

- Hydroxyethylcellulose[28] wird durch Umsetzung einer alkalisch vorbehandelten Cellulose mit Ethylenoxid hergestellt. Sie wird analog wie Hydroxypropylcellulose eingesetzt, ist aber (bei gleichem Substitutionsgrad) etwas hydrophiler als diese.
- Carboxymethylcellulosen wird durch Umsetzung von alkalisch vorbehandelter Cellulose mit Chloressigsäure hergestellt. Sie hat ein sehr breites Anwendungsspektrum, so ist sie z. B. als Lebensmittelzusatzstoff zugelassen und hat die Nummer E 466, dort wird es als Verdickungsmittel und zur Verbesserung der Konsistenz eingesetzt. In der Pharmazie nutzt man sie als Tablettensprengmittel.[29]
- Diethylaminoethylcellulose wird durch Umsetzung von alkalisch vorbehandelter Cellulose mit 3-Chlortriethylamin hergestellt.[30] Sie wird als schwach basischer Ionenaustauscher verwendet, speziell für die Trennung von Proteinen.[31]

Bei der Herstellung von Hydroxypropylmethylcellulose, Hydroxypropylcellulose und Hydroxyethylcellulose kann es immer zur Bildung von mehrgliedrigen Seitenketten aus Polyethylenoxid bzw. Polypropylenoxid kommen, noch bevor alle OH-Gruppen der Cellulose substituiert sind. Reaktionstechnisch lässt sich nicht vermeiden, dass ein relativ uneinheitliches Produkt entsteht.
Stärkederivate Bearbeiten
Im Gegensatz zu Cellulose ist Stärke und viele ihrer Derivate von Menschen verdaubar und daher gibt es eine Vielzahl von Stärkederivaten, die in großem Maß in der Lebensmitteltechnologie zur Modifikation von Lebensmitteln.[32][33] sowie bei der Papierherstellung[34] eingesetzt werden. Die Verdaulichkeit nimmt mit steigendem Substitutionsgrad allerdings ab und einige sehr hoch substituierte Derivate sind unverdaulich. Meist wird keine native, sondern oxidativ oder enzymatisch abgebaute Stärke eingesetzt, weil die Molmassen nativer Stärken speziell bei Amylopektinen oft so hoch sind, dass die Löslichkeit schlecht, oder die Lösungsviskositäten sehr hoch sind, dass Derivatisierungen stark erschwert werden.
Kationische Stärke Bearbeiten
Kationische Stärke wird in großem Umfang für die Herstellung von Papier eingesetzt. Dort dient sie u. a. als Retentionsmittel und zur Trockenverfestigung.[35] Aufgesprühte kationische Stärke verbessert die Bedruckbarkeit.[34] Im Gegensatz zu anderen Stärkederivaten haben kationische Stärken einen sehr niedrigen Substitutionsgrad, der typischerweise zwischen 0,03 und 0,1 liegt.[36]

Stärkeester Bearbeiten
- Acetylierte Stärke (E 1420) wird durch Umsetzung von Stärke mit Essigsäureanhydrid hergestellt. E 1420 bildet klare und stabile Lösungen und wird zur Stabilisierung von Tiefkühllebensmittel und Milcherzeugnissen verwendet.[37]

Da bei dieser Reaktion nicht alle Hydroxygruppen reagieren, entstehen Gemische mit unterschiedlich hohem Substitutionsgrad. Auch der Substitutionsgrad der einzelnen Stärkebausteine innerhalb eines Polymers kann unterschiedlich hoch ausfallen. Analoge Gemische entstehen bei den folgenden Reaktionen.
- Stärkesulfate werden durch die Umsetzung von alkalisch vorbehandelter Stärke mit Chlorsulfonsäure hergestellt. Sie waren eine Zeit lang als Substitut für Heparin im Gespräch.[38]
- Stärkenitrate werden durch Umsetzung von Stärke mit konzentrierter Schwefelsäure und Salpetersäure hergestellt. Sie haben ähnliche Eigenschaften wie Cellulosenitrate, aber eine weit geringere technische und wirtschaftliche Bedeutung.[39]
- Stärkexanthogenate werden durch die Umsetzung alkalisch vorbehandelter Stärke mit Kohlenstoffdisulfid hergestellt. Sie werden in der Papierindustrie zur Papierverfestigung und zur Herstellung von Elastomeren verwendet.[38]
- Stärkecitrate werden durch Umsetzung von Stärke mit Citronensäure hergestellt.[40] Sie werden in der Lebensmitteltechnologie bei Tiefkühlware eingesetzt.[38]
- Stärkesuccinate werden durch Umsetzung von Stärke mit Bernsteinsäureanhydrid hergestellt.[41] Sie erweisen sich sowohl als gute Stabilisatoren als auch als gute Emulgatoren und werden zur Aromastabilisierung von Lebensmitteln vorgeschlagen.[38]
- Stärkephosphate werden durch Umsetzen von Stärke mit Mononatriumorthophosphat oder Dinatriumorthophosphat hergestellt.[42][43] Sie werden speziell bei säurehaltigen Lebensmitteln, die stark erwärmt (sterilisiert) werden, eingesetzt.[44]
- Stärkenatriumoctenylsuccinat (E 1450) wird durch Umsetzung von Stärke mit Octenylbernsteinsäureanhydrid hergestellt.[45] Sie quillt bereits in kaltem Wasser und wirkt als Emulgator, der Wasser/Öl-Emulsionen stabilisiert. Zudem bildet es stabile, gefrierstabile Schäume.[46]
Stärkeether Bearbeiten
- Hydroxypropylstärke wird durch die Umsetzung von alkalisch vorbehandelter Stärke mit Propylenoxid hergestellt.[47] Sie wird als hitzestabiles Verdickungsmittel verwendet, speziell für Nahrungsmittel, die sterilisiert werden.[48]

- Hydroxyethylstärke wird durch die Umsetzung von alkalisch vorbehandelter Stärke mit Ethylenoxid hergestellt. Sie wird für die Papierherstellung und als Textilhilfsstoff verwendet.[49] Bis 2013 auch als Plasmaersatzstoff, momentan ist es aber für diesen Zweck nicht mehr zugelassen.[50]

- Carboxymethylstärke wird durch die Umsetzung von alkalisch vorbehandelter Stärke mit Chloressigsäure hergestellt.[51] Sie bildet hochviskose Lösungen, ohne Gelbildung[34] und ist ein Grundstoff für abbaubare Tenside.[52]

Posttranslationale Modifikationen Bearbeiten
Posttranslationale Proteinmodifikationen (PTM) sind Veränderungen von Proteinen, die nach der Translation stattfinden. Auf diesem Weg können Aminosäuren in Proteine eingebaut werden, die kein eigenes Kodon besitzen. So besitzt Hydroxyprolin kein Kodon und kann nicht direkt in Proteine eingebaut werden, sondern wird in Kollagen durch Prolyl-4-Hydroxylase aus Prolin hergestellt.

Posttranslationale Modifikation lassen sich in folgende Gruppen einteilen
- Abspaltungen
- Einfügung von Anorganischen Gruppen
- Einfügung von Organischen Gruppen
- Einfügung von Lipidgruppen (als Sonderfall)
- Einfügen von Bindungen
- Bindung an größere Moleküle
- Veränderung einzelner Aminosäuren
- Andere Reaktionen
Bei dieser Einteilung gibt es allerdings Überschneidungen und Uneindeutigkeiten, weil es eine empirische und nicht streng systematische Einteilung ist und nicht alle Posttranslationale Modifikationen Polymeranaloge Reaktionen sind.[53]
Ohne Posttranslationale Modifikationen könnten viele Proteine ihre Aufgaben nicht erfüllen, weil sie sonst eine andere als die geforderte Konfiguration hätten, zu hydrophil, oder hydrophob wären, oder andere Eigenschaften nicht erfüllten. Die meisten Posttranslationale Modifikationen sind enzymkatalysierte Reaktionen und keine, die durch DNA/RNA gesteuert werden. Sie können an unterschiedlichen Stellen der Zellen stattfinden, nicht nur in den Ribosomen.
DNA Bearbeiten
Zur Kontrolle der Genexpression wird DNA in Lebewesen chemisch modifiziert. Eine sowohl in Pro- als auch Eukaryoten vorkommende Variante davon ist die Methylierung von Cytosin durch ein Enzym aus der Gruppe der DNA-Methyltransferasen. Das Enzym überträgt eine Methylgruppe von S-Adenosylmethionin (SAM) auf Cytosin (hier dargestellt an einer freien Pyrimidinbase):

Dabei entstehen S-Adenosylhomocystein (SAH) und 5-Methylcytosin. Mit den Folgen dieser und anderer chemischer Modifikationen am Genom beschäftigt sich die Epigenetik.[54]
Andere natürliche Polymere Bearbeiten
Chitosan wird aus Chitin durch Verseifung oder enzymatischer Deacetylierung hergestellt. Auch Chitosan hat eine sehr breite Anwendung.[55]

Abgrenzung von Polymeranalogen Reaktionen zur Vernetzung Bearbeiten
Vulkanisation Bearbeiten
Das Vulkanisieren (Vernetzen) von Kautschuk zu Gummi zählt nicht zu den Polymeranalogen Reaktionen, sondern zu den Vernetzungen, weil die Molmasse des vulkanisierten Produktes um ein Vielfaches höher ist als die des Eduktes.[56] Dies ist ein Beispiel, dass ein Polymer mit einem niedermolekularen Vernetzer (Schwefel) zu einem Netzwerk reagiert.

Schematische Präsentation von zwei Polyisoprenketten (blau und grün) nach der Vulkanisation mit Schwefel (n = 0, 1, 2, 3 …). Die Polyisoprenketten sind hier über zwei Schwefelbrücken miteinander verknüpft
Mischsysteme Bearbeiten
Es gibt Systeme, bei denen sowohl polymeranaloge Reaktionen, als auch Vernetzungen stattfinden. Beispiele sind die Herstellung von Polyamidoamin-epichlorhydrinharzen und die Herstellung von Kohlenstofffasern. Bei den Stärkeestern mehrbasiger Säuren liegt je nach Stöchiometrie eine Polymeranaloge Reaktion oder eine mehr oder weniger stark ausgeprägte Vernetzung vor.
Stärkeester mehrbasiger Säuren Bearbeiten
Distärkephosphat[57] und Phosphatiertes Distärkephosphat[58] gehören zu den teilweise vernetzten Polymeren, weil die Phosphatgruppen mehrere Ketten miteinander verbinden können. Auch die Stärkesuccinate und Stärkeadipate gehören zu den teilweise vernetzten Polymeren.[38]
Herstellung von Polyamidoamin-epichlorhydrinharzen Bearbeiten
Polyamidoamin-epichlorhydrinharze werden u. a. als Nassfestmittel bei der Papierproduktion verwendet. Hier wird aus Adipinsäure und Diethylentriamin (oder anderen Polyaminen) durch Polykondensation ein Prepolymer hergestellt, das in einer polymeranalogen Reaktion mit Epichlorhydrin zu einem reaktiven Prepolymer umgesetzt wird, das anschließend vernetzt werden kann.[59] Dies ist ein Beispiel, bei dem reaktive Polymere miteinander zu einem Netzwerk reagieren.

Herstellung von Kohlenstofffasern Bearbeiten
Kohlenstofffasern werden zum größten Teil aus Polyacrylnitril (PAN) hergestellt. Dazu wird PAN gesponnenen und verstreckt und diese Fasern in einer polymeranalogen Reaktion zu einem Leiterpolymer umgesetzt. Diese Vorreaktion verläuft in zwei Schritten. Im ersten werden unter sauerstofffreien Bedingungen die CN-Gruppen bei 200–300 °C cyclisiert und in einem zweiten Schritt durch Oxidation mit Sauerstoff dieses Polymer aromatisiert. In einem weiteren Schritt wird es unter Eliminierung von HCN oder Stickstoff graphitisiert, = vernetzt[60] Die folgenden Abbildungen geben einen schematisierten und idealisierten Ablauf der Reaktionen an. Zur besseren Anschaulichkeit wurde die lineare Nitrilgruppe gewinkelt dargestellt.


Einzelnachweise Bearbeiten
- ↑ a b Hans-Georg Elias: Makromoleküle. Band 1, 6. Auflage, Wiley, Weinheim 1999, ISBN 3-527-29872-X, S. 554 ff.
- ↑ Beschreibung des Beizens von Naturfasern, abgerufen am 17. Dezember 2017.
- ↑ Jochen Gartz: Vom Griechischen Feuer zum Dynamit – eine Kulturgeschichte der Explosivstoffe. E.S. Mittler & Sohn, Hamburg / Berlin / Bonn 2007, ISBN 978-3-8132-0867-2.
- ↑ Victor Emmanuel Yarsley: Über die Herstellung und physikalischen Eigenschaften der Celluloseacetate. Julius Springer Verlagsbuchhandlung, Berlin 1927, S. 5, doi:10.1007/978-3-642-98939-1.
- ↑ Positionspapiere der DECHEMA abgerufen am 28. September 2020.
- ↑ PDF des Positionspapiers Polymerisationstechnik – eine kritische Bestandsaufnahme mit Ausblick abgerufen am 28. September 2020.
- ↑ a b Sebastian Koltzenburg, Oskar Nuyken, Michael Maskos: Polymere: Synthese, Eigenschaften und Anwendungen. 1. Auflage. Springer Spektrum, Berlin Heidelberg 2014, ISBN 978-3-642-34772-6, doi:10.1007/978-3-642-34773-3 (ISBN 978-3-642-34773-3 (E-Book)).
- ↑ Hans Rudolf Christen, Fritz Vögtle: Organische Chemie. Band 1, Salle + Sauerländer, Frankfurt am Main 1988, ISBN 3-7935-5397-3, S. 132 und 436.
- ↑ Hans-Georg Elias: Makromoleküle. Band 4, 6. Auflage, Wiley, Weinheim 1999, ISBN 978-3-527-29962-1, S. 208 ff.
- ↑ Produktliste teilverseifter Polyvinylalkohole der Firma Kuraray. Abgerufen am 25. März 2016.
- ↑ Karl Oberbach (Hrsg.): Saechtling Kunststoff-Taschenbuch. Carl Hanser Verlag, München / Wien 2004, ISBN 3-446-22670-2, S. 458.
- ↑ I. Stolkin, T.-K. Ha, Hs. H. Günthard: N-methylmethyleneimine and ethylideneimine: Gas- and matrix-infrared spectra, AB initio calculations and thermodynamic properties. In: Chemical Physics. Band 21, Nr. 3, 1977, S. 327–347, doi:10.1016/0301-0104(77)85189-6.
- ↑ Blandine Brissault, Antoine Kichler, Christine Guis, Christian Leborgne, Olivier Danos, Hervé Cheradame: Synthesis of Linear Polyethylenimine Derivatives for DNA Transfection. In: Bioconjugate Chemistry. Band 14, Nr. 3, 2003, S. 581–587, doi:10.1021/bc0200529.
- ↑ a b Hans-Georg Elias: Makromoleküle. Band 1, 6. Auflage, Wiley, Weinheim 1999, ISBN 3-527-29872-X, S. 558–559.
- ↑ a b c M. D. Lechner, K. Gehrke und E. H. Nordmeier: Makromolekulare Chemie, 4. Auflage, Birkhäuser Verlag, 2010, S. 480, ISBN 978-3-7643-8890-4.
- ↑ Ionenaustausch für Laien, eine Beschreibung der Firma Rohm und Haas (Memento vom 27. August 2010 im Internet Archive). Abgerufen am 15. März 2016.
- ↑ a b Hans-Georg Elias: Makromoleküle. Band 1, 6. Auflage, Wiley, Weinheim 1999, ISBN 3-527-29872-X, S. 564.
- ↑ Robert Bruce Merrifield: Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. In: Journal of the American Chemical Society. Band 85, Nr. 14, 1963, S. 2149–2154, doi:10.1021/ja00897a025.
- ↑ Erich Wünsch: Synthese von Peptid-Naturstoffen: Problematik des heutigen Forschungsstandes. In: Angewandte Chemie. Band 83, Nr. 20, 1971, S. 773–782, doi:10.1002/ange.19710832002.
- ↑ Hans-Georg Elias: Makromoleküle. Band 1, 6. Auflage, Wiley, Weinheim 1999, ISBN 3-527-29872-X, S. 561.
- ↑ Eintrag zu Acetatseide. In: Römpp Online. Georg Thieme Verlag, abgerufen am 11. März 2016.
- ↑ Tischtennisregeln A, Punkt 3.3 (Memento vom 13. Februar 2007 im Internet Archive). In: tischtennis.de. Abgerufen am 11. März 2016.
- ↑ Eintrag zu Celluloid. In: Römpp Online. Georg Thieme Verlag, abgerufen am 18. März 2016.
- ↑ Eintrag zu Hydroxypropylcellulosen. In: Römpp Online. Georg Thieme Verlag, abgerufen am 11. März 2016.
- ↑ Technisches Merkblatt Metylan Normal (Memento vom 11. März 2016 im Internet Archive). Henkel, S. 3, abgerufen am 11. März 2016 (PDF; 265 kB).
- ↑ Aqualon Ethylcellulose (Memento vom 16. Mai 2011 im Internet Archive) auf der Seite der Ashland Inc., abgerufen am 14. März 2016.
- ↑ Eintrag zu Verdickungsmittel. In: Römpp Online. Georg Thieme Verlag, abgerufen am 11. März 2016.
- ↑ Eintrag zu Hydroxyethylcellulosen. In: Römpp Online. Georg Thieme Verlag, abgerufen am 31. März 2016.
- ↑ Verschiedene Anwendungen von Carboxymethylcellulose, abgerufen am 14. März 2016.
- ↑ Ronald W. Rousseau, James K. Ferrell, Robert F. Reardon: Synthesis of diethylaminoethyl cellulose on cotton fabric. In: Industrial & Engineering Chemistry Product Research and Development. 23. Jahrgang, Nr. 2, 1. Juni 1984, ISSN 0196-4321, S. 250–252, doi:10.1021/i300014a015.
- ↑ Datenblatt 2,3-Diethylaminoethylcellulose bei Sigma-Aldrich, abgerufen am 14. März 2016 (PDF).
- ↑ Stärkederivate für die Lebensmittelindustrie I, abgerufen am 15. März 2016.
- ↑ Stärkederivate für die Lebensmittelindustrie II, abgerufen am 15. März 2016.
- ↑ a b c Übersicht über kationische Stärken, die in der Papierherstellung verwendet werden, abgerufen am 15. März 2016.
- ↑ Mano Wolf: Stärkeeinsatz im Papier und deren Dosiereinrichtungen (Memento vom 24. März 2016 im Internet Archive). Abgerufen am 19. März 2016.
- ↑ Fa. Yumpu Stärkeeinsatz im Papier, abgerufen am 29. September 2017.
- ↑ Eintrag zu Stärkeacetat. In: Lexikon der Ernährung. Spektrum der Wissenschaft Verlag, abgerufen am 15. März 2016.
- ↑ a b c d e Eintrag zu Stärkeester. In: Römpp Online. Georg Thieme Verlag, abgerufen am 18. März 2016.
- ↑ Eintrag zu Stärkenitrate. In: Römpp Online. Georg Thieme Verlag, abgerufen am 18. März 2016.
- ↑ H. Klaushofer, E. Berghofer, W. Steyrer: Stärkecitrate – Produktion und anwendungs‐technische Eigenschaften. In: Starch, 1978, Vol. 30, Nr. 2, S. 47–51, doi:10.1002/star.19780300204.
- ↑ Frank Böttger: Synthese und Charakterisierung neuer Stärkederivate für die klinische Anwendung. Kassel, 2003, S. 29, https://kobra.uni-kassel.de/handle/123456789/700.
- ↑ Eintrag zu Stärkephosphate. In: Römpp Online. Georg Thieme Verlag, abgerufen am 18. März 2016.
- ↑ Eintrag zu Monostärkephosphat. In: lebensmittellexikon.de Frank Massholder, abgerufen am 15. März 2016.
- ↑ Eintrag zu Monostärkephosphat. In: lebensmittellexikon.de Frank Massholder, abgerufen am 15. März 2016.
- ↑ Joint FAO/WHO Expert Committee on Food Additives (JECFA), Monograph für STARCH SODIUM OCTENYL SUCCINATE, abgerufen am 9. Dezember 2014.
- ↑ Eintrag zu Stärkenatriumoctenylsuccinat. In: lebensmittellexikon.de Frank Massholder, abgerufen am 15. März 2016.
- ↑ Eintrag zu Hydroxypropylstärken. In: Römpp Online. Georg Thieme Verlag, abgerufen am 4. Mai 2020.
- ↑ Eintrag zu Hydroxypropylstärke. In: lebensmittellexikon.de Frank Massholder, abgerufen am 15. März 2016.
- ↑ Eintrag zu Hydroxyethylstärken. In: Römpp Online. Georg Thieme Verlag, abgerufen am 18. März 2016.
- ↑ Hydroxyethylstärke: EMA für Verbot von Volumenersatzmittel. In: aerzteblatt.de. Deutsches Ärzteblatt, 14. Juni 2013, abgerufen am 18. März 2016.
- ↑ Prof. Dr. M. Čeh Dipl.‐Ing.: Darstellung der Carboxymethylstärke. In: Starch, 1972, Vol. 24, Nr. 4, S. 124–127, doi:10.1002/star.19720240406.
- ↑ Eintrag zu Stärkeether. In: Römpp Online. Georg Thieme Verlag, abgerufen am 18. März 2016.
- ↑ Joachim Rassow et al.: Biochemie. Thieme Verlag, Stuttgart 2008, ISBN 978-3-13-125352-1, Kapitel 12: Genexpression: 12.7 Co- und posttranslationale Modifikation von Proteinen, doi:10.1055/b-0034-88959.
- ↑ Alfred Nordheim, Rolf Knippers (Hrsg.): Molekulare Genetik. 10. Auflage. Thieme, Stuttgart 2015, ISBN 978-3-13-477010-0, S. 451 f.
- ↑ Chitosan – ein Überblick. (Memento vom 1. November 2013 im Internet Archive) Hochschule Emden/Leer, abgerufen am 14. März 2016.
- ↑ Beschreibung der Vulkanisation auf der Seite von Prof. Blume, abgerufen am 14. März 2016.
- ↑ Eintrag zu Distärkephosphat. In: lebensmittellexikon.de Frank Massholder, abgerufen am 15. März 2016.
- ↑ Eintrag zu Phosphatiertes Distärkephosphat. In: lebensmittellexikon.de Frank Massholder, abgerufen am 15. März 2016.
- ↑ Andreas Pingel-Keuth: Papierproduktion: Von Zellstoff zu Filtertüte, Schreibpapier, … In: Chemie in unserer Zeit. Band 39, Nr. 6, 2005, S. 402–409, doi:10.1002/ciuz.200500234.
- ↑ Präsentation der Firma Toho Tenax zu Eigenschaften und Herstellung von Kohlenstofffasern S. 2, abgerufen am 5. September 2017.