Plumbylene
Plumbylene (oder Plumbylidene) sind divalente Blei(II)organyle und Tetrylene mit der generellen chemischen Formel R2Pb. Plumbylene haben sechs Elektronen in ihrer Valenzschale und sind niedervalente Verbindungen.

Das erste beschriebene Plumbylen war das Dialkylplumbylen [(Me3Si)2CH]2Pb, welches 1973 von Michael Lappert und Mitarbeitern synthetisiert wurde.[1]
Plumbylene können als kohlenstoffsubstituierte Plumbylene, Plumbylene, die durch ein Gruppe 15 oder 16 Element stabilisiert werden, und als monohalogenierte Plubylene klassifiziert werden.[2]
Synthese Bearbeiten
Plumbylene können zum einen durch Transmetallierung von Bleidihalogeniden mit lithiumorganischen Verbindungen oder Grignardreagenzien synthetisiert werden.[2] Das erste Plumbylen [((CH3)3Si)2CH]2Pb von Michael Lappert et al. wurde entsprechend durch die Reaktion von Bleidichlorid mit [((CH3)3Si)2CH]Li erhalten.[1] Die äquimolare Zugabe von Lithiumorganylen zu Bleidihalogeniden führt zu monohalogenierten Plumbylenen, die Zugabe von zwei Äquivalenten Lithiumorganylen zu diorganylsubstituierten Plumbylenen.[3] Auf diese Weise können Dialkyl-,[1] Diaryl-,[4] Diamido-,[5] Dithioplumbylene[3] erhalten werden. Die Zugabe einer Organolithium- oder Grignard-Verbindung mit einem unterschiedlichen organischen Substituenten zu monohalogenierten Plumbylenen führt zum Erhalt von heteroleptischen Plumbylenen.[3]

Auch die Transmetallierung von [((CH3)3Si)2N]2Pb kann zur Synthese von Diarylplumbylenen,[6] Disilylplumbylenen,[7] und gesättigten N-heterocyclischen Plumbylenen[8] verwendet werden.
Alternativ können Plumbylene auch durch die reduktive Dehalogenierung von tetravalenten Organobleiverbindungen (R2PbX2).[6]

Struktur und Bindung Bearbeiten

Ein Kernaspekt der elektronischen Struktur von Plumbylenen und damit ihrer Reaktivität hängt mit dem Inert-Pair-Effekt zusammen. So führt der wachsende Abstand zwischen den Energien der s- und p-Orbitale bei zunehmender Ordnungszahl der Elemente der Gruppe 14 zusammen mit einer starken relativistischen Kontraktion des 6s-Orbitals nur zu einem geringen Hybridisierungsgrad der s- und p-Orbitale. In der Folge liegt das 6s-Orbital energetisch sehr niedrig und ist verhältnismäßig inert.[9] Das führt in der Konsequenz dazu, dass Plumbylene einen Singulettzustand einnehmen, eine hohe Energiedifferenz zwischen Singulett- und Triplett-Zustand aufweisen und in Lösung zu einem Gleichgewicht zwischen monomerer und dimerer Form neigen (siehe unten).[9] Das steht im Gegensatz zu Carbenen, die häufig einen Triplettgrundzustand aufweisen und leicht zu Alkenen dimerisieren.

In Dimethylblei (CH3)2Pb beträgt die the Pb–C-Bindungslänge 2.267 Å und der C–Pb–C-Bindungswinkel 93.02°. Der Singulett-Triplett-Abstand beträgt 36,99 kcal mol−1.[10]
Verschiedene Visualisierungsmethoden wie Kohn-Sham-Orbitale oder NBOs zeigen bei Diphenylblei Ph2Pb eine deutliche Lokalisation eines freien Elektronenpaares in Form des höchsten besetzten Molekülorbitals (HOMO) am Bleizentrum. Ebenso findet sich dort das niedrigste unbesetzte Molekülorbital (LUMO).[11]
-
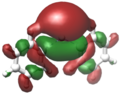 HOMO von Ph2Pb
HOMO von Ph2Pb -
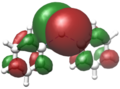 LUMO von Ph2Pb
LUMO von Ph2Pb -
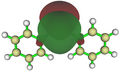
 Das mittels NBO berechnete freie 6s-Elektronenpaar
Das mittels NBO berechnete freie 6s-Elektronenpaar -
 Das mittels NBO berechnete vakante 6p-Orbital
Das mittels NBO berechnete vakante 6p-Orbital




Die Pb–C-Bindungslängen betragen in dieser Verbindung 2.303 Å und der C–Pb–C-Bindungswinkel 105.7°. Ungeachtet von verschiedenen theoretischen Rechenniveaus kann der größere Bindungswinkel in Diphenylblei gegenüber Dimethylblei auf die Abstoßung der sterisch anspruchsvolleren Phenylgruppen zurückgeführt werden.
Plumbylene treten als reaktive Intermediate bei der Bildung von tetravalenten Plumbanen (R4Pb) auf.[12] Obwohl der Inert-Pair-Effekt darauf deutet, dass die divalenten Spezies thermodynamisch stabiler sein sollten, kann die Abwesenheit von stabilisierenden Substituenten zu einer Sensitivität gegenüber Licht und Wärme und damit zu Polymerisation und Disproportionierung führen. Entsprechend entsteht bei einer solchen Reaktion auch elementares Blei.[12][13]
Plumbylene können als Monomere durch die Verwendung von sterisch anspruchsvollen Liganden (kinetische Stabilisierung) oder hetreroatomhaltigen Substituenten, die Elektronendichte in das vakante 6p-Orbital donieren (thermodynamische Stabilisierung), stabilisiert werden.[2]
Elektronische Stabilisierung von Plumbylenen Bearbeiten
Plumbylene können durch funktionelle Gruppen stabilisiert werden, die Elektronendichte in das vakante 6p-Orbital des Bleis donieren. Die geläufigsten intramolekularen Wege sind zum einen die Einführung einer funktionellen Gruppe mit einem dem Blei benachbarten Atom, das ein freies Elektronenpaar besitzt, oder die Koordination einer Lewis-basischen Funktionalität aus einer anderen Stelle des Moleküls.[14]
Ein Beispiel für den ersten Fall sind dem Blei benachbarte Elemente der Gruppen 15 und 16, analog zu Fischer-Carbenen oder N-heterocyclischen Carbenen.[2][4][15][16]
_Valence_orbital_diagram_showing_the_donation_of_a_lone_pair_on_heteroatom_E_to_the_vacant_6p_orbital_of_adjacent_Pb.png)
Beispiele für den zweiten Fall sind zum Beispiel koordinierende Aminogruppen aus dem Rückgrat des Moleküls.[14] Allerdings gibt es sogar Beispiele für die Koordination von Fluoratomen aus Trifluormethylgruppen.[17]
-
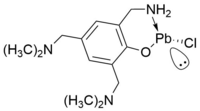 Stabilisierung eines Plumbylens durch die Koordination einer Amingruppe eines Substituenten
Stabilisierung eines Plumbylens durch die Koordination einer Amingruppe eines Substituenten -
![Lewisstruktur von [2,4,6-(CF3)3C6H2]2Pb](//upload.wikimedia.org/wikipedia/commons/thumb/2/2c/%282%2C4%2C6-%28CF3%293C6H2%292Pb_skeletal.png/200px-%282%2C4%2C6-%28CF3%293C6H2%292Pb_skeletal.png) Lewisstruktur von [2,4,6-(CF3)3C6H2]2Pb
Lewisstruktur von [2,4,6-(CF3)3C6H2]2Pb -
![Molekülstruktur von [2,4,6-(CF3)3C6H2]2Pb im Kristall](//upload.wikimedia.org/wikipedia/commons/thumb/d/dc/%282%2C4%2C6-%28CF3%293C6H2%292Pb_X-ray.png/186px-%282%2C4%2C6-%28CF3%293C6H2%292Pb_X-ray.png) Molekülstruktur von [2,4,6-(CF3)3C6H2]2Pb im Kristall
Molekülstruktur von [2,4,6-(CF3)3C6H2]2Pb im Kristall
.png)
![Lewisstruktur von [2,4,6-(CF3)3C6H2]2Pb](/wiki/Datei:(2,4,6-(CF3)3C6H2)2Pb_skeletal.png)
![Molekülstruktur von [2,4,6-(CF3)3C6H2]2Pb im Kristall](/wiki/Datei:(2,4,6-(CF3)3C6H2)2Pb_X-ray.png)
Agostische Wechselwirkungen Bearbeiten
Auch agostische Wechselwirkungen können Plumbylene stabilisieren. So zeigten computerchemische Untersuchungen von {[R(Me3)2Si][Me2P(BH3)]CH}2Pb (R = Me oder Ph), dass die Wechselwirkungen von bindenden B–H-Orbitalen mit dem vakanten 6p-Orbital die Energie des Moleküls um etwa 38 kcal mol−1 senken. Diese Beobachtung wurde durch Kristallstrukturanalysen untermauert, in denen die B–H Bindungen nur einen geringen Abstand zum Blei aufweisen.[18]
-
![Frontansicht von {(Me3Si)[Me2P(BH3)]CH}2Pb](//upload.wikimedia.org/wikipedia/commons/thumb/7/70/Plumbylene_agostic_front_view2.png/152px-Plumbylene_agostic_front_view2.png) Frontansicht von {(Me3Si)[Me2P(BH3)]CH}2Pb
Frontansicht von {(Me3Si)[Me2P(BH3)]CH}2Pb -
![Draufsicht auf {(Me3Si)[Me2P(BH3)]CH}2Pb](//upload.wikimedia.org/wikipedia/commons/thumb/f/f8/Plumbylene_agostic_top_view2.png/191px-Plumbylene_agostic_top_view2.png) Draufsicht auf {(Me3Si)[Me2P(BH3)]CH}2Pb
Draufsicht auf {(Me3Si)[Me2P(BH3)]CH}2Pb
![Frontansicht von {(Me3Si)[Me2P(BH3)]CH}2Pb](/wiki/Datei:Plumbylene_agostic_front_view2.png)
![Draufsicht auf {(Me3Si)[Me2P(BH3)]CH}2Pb](/wiki/Datei:Plumbylene_agostic_top_view2.png)
Reaktivität Bearbeiten
Wie oben erwähnt tendieren unstabilisierte Plumbylene zu Polymerisation, Disproportionierung oder Dimerisierung. Allerdings gibt es noch eine Vielzahl weiterer Reaktivitäten.
Bildung von Lewis-Säure-Base-Addukten Bearbeiten
So verhalten sich Plumbylene durch das vakante 6p-Orbital Lewis-sauer und neigen entsprechend dazu, Addukte mit Lewisbasen wie Trimethylaminoxid,[19] 1-Azidoadamantan (AdN3)[20] oder Mesitylazid (MesN3)[19] zu bilden. Im Gegensatz dazu führt die Reaktion von Stannylenen und Trimethylaminoxid durch eine Oxidation des Zinns von der Oxidationsstufe +II auf +IV zum korrespondierenden Zinnoxid. Das lässt sich auf den deutlich schwächeren Inert-Pair-Effekt des Zinns und einer damit verbundenen Vorliebe zur Oxidation zurückführen.[21]
Dimerisierung Bearbeiten

Plumbylene können auf zwei Wegen dimersieren. Zum einen durch die Bildung von Pb=Pb-Doppelbindungen und damit der Bildung von Diplumbylenen, zum anderen aber auch verbrückt durch Halogen–Blei-Wechselwirkungen. Nichthalogenierte Plumbylene liegen in Lösung in einem Gleichgewicht zwischen monomerer und dimerer Form vor. Im Festkörper können sie aufgrund von geringen Dimerisierungsenergien sowohl als monomere oder als dimere Vorliegen. Das ist in der Regel vom sterischen Anspruch der Substituenten abhängig.[2][9][22][23] Allerdings große Substituenten auch zu einer sterischen Verhinderung der Dimerisierung und damit zum reinen Vorliegen des Monomers führen.[3][23][24]

Die Triebkraft der Dimerisierung lässt sich auf die amphotere Lewisverhalten von Plumbylenen zurückführen. So kann das freie 6s-Elektronenpaar als Lewisbase und Elektronendonor dienen, wohingegen das vakante 6p-Orbital als Lewissäure und Elektronenakzeptor fungieren kann.[7][11] Diplumbylene weisen in der Folge eine trans-bent-Struktur, ähnlich zu den leichteren Homologen (Disilylene, Digermylene, Distannylene) auf.[9] Dabei sind die beobachteten Pb–Pb-Bindungen (2,90–3,53 Å) tendenziell länger als in Diblumbanen R3PbPbR3 (2,84–2,97 Å).[23] Dieses kontraintuitive Verhalten lässt sich auf zwei 6s-6p-Donor-Akzeptor-Wechselwirkungen zurückführen, die energetisch günstiger sind als der Überlapp von spn-Orbitalen mit einem höheren Hybridisierungsgrad.[23]
In monohalogenierten Plumbylenen ist außerdem das Halogenatom dazu in der Lage Elektronendichte in das vakante 6p-Orbital an einem Bleiatom eines zweiten Plumbylens zu donieren, wodurch verbrückte Spezies entstehen. Solche monohalogenierten Plumbylene liegen im Allgemeinen in Lösung monomer und im Festkörper als Dimer vor, allerdings kann auch hier ein hoher sterischer Anspruch der Substituenten die Dimerisierung verhindern.[2] Da die Dimerisierungsenergie von Tetrylenen in der Gruppe 14 mit steigender Ordnungszahl des Zentralatoms abnimmt, dimerisieren monohalogenierte Stannylene und Plumbylene eher verbrückend als monohalogenierte Silylene und Plumbylene. Letztere dimersieren eher über die oben beschriebenen Doppelbindungen.[2]

Insertionsreaktionen Bearbeiten
Ähnlich zu Carbenen[25] und den anderen Tetrylenen[2] können Plumbylene Insertionsreaktionen eingehen. Insbesondere sind Insertionen in C–X- (X = Br, I) und E–E-Bindungen der Gruppe 16 (E = S, Se) bekannt.[6]

Transmetallierung Bearbeiten
Plumbylene können weiterhin nukleophile Substitutionsreaktionen mit metallorganischen Reagenzien eingehen. So begehen sie zum Beispiel mit geeigneten Gruppe 13-Verbindungen entsprechende Metathesen.[24]

Plumbylene mit unterschiedlichen Substituenten können ebenso Transmetallierungsreaktionen eingehen und ihre Substituenten austauschen. Die Triebkraft dieser Reaktion ist die geringere sterische Spannung im Produkt bei einer gleichzeitig geringen Dissoziationsenergie der Pb–C-Bindung.[26]
Anwendung Bearbeiten
2.png)
Plumbylene können außerdem als σ-Donor-σ-Akzeptorliganden in Metallkomplexen eingesetzt werden. Die σ-Donorfuktionalität resultiert dabei aus dem besetzten 6s-Orbital und die σ-Akzeptorfunktionalität aus dem unbesetzten 6p-Orbital.
Bei Raumtemperatur stabile Plumbylene können außerdem als Precursoren in der chemischen Gasphasenabscheidung oder der Atomlagenabscheidung eingesetzt werden, um bleihaltige Materialien zu erhalten.[27] Dithioplumbylene und Dialkoxyplumbylene könnten außerdem sinnvolle Ausgangsmaterialien für den Halbleiter Blei(II)-sulfid beziehungsweise das piezokeramische Blei-Zirkonat-Titanat sein.[28]
Einzelnachweise Bearbeiten
- ↑ a b c Peter J. Davidson, Michael F. Lappert: Stabilisation of metals in a low co-ordinative environment using the bis(trimethylsilyl)methyl ligand; coloured Sn II and Pb II alkyls, M[CH(SiMe3)2]2. In: Journal of the Chemical Society, Chemical Communications. Nr. 9, 1973, ISSN 0022-4936, S. 317a, doi:10.1039/c3973000317a.
- ↑ a b c d e f g h Yoshiyuki Mizuhata, Takahiro Sasamori, Norihiro Tokitoh: Stable Heavier Carbene Analogues. In: Chemical Reviews. Band 109, Nr. 8, 12. August 2009, ISSN 0009-2665, S. 3479–3511, doi:10.1021/cr900093s.
- ↑ a b c d Lihung Pu, Brendan Twamley, Philip P. Power: Terphenyl Ligand Stabilized Lead(II) Derivatives of Simple Organic Groups: Characterization of Pb(R)C6H3-2,6-Trip2 (R = Me, t-Bu, or Ph; Trip = C6H2-2,4,6-i-Pr3), {Pb(μ-Br)C6H3-2,6-Trip2}2, py·Pb(Br)C6H3-2,6-Trip2 (py = Pyridine), and the Bridged Plumbylyne Complex [{W(CO)4}2 (μ-Br)(μ-PbC6H3-2,6-Trip2)]. In: Organometallics. Band 19, Nr. 15, 1. Juli 2000, ISSN 0276-7333, S. 2874–2881, doi:10.1021/om0001624.
- ↑ a b David H. Harris, Michael F. Lappert: Monomeric, volatile bivalent amides of group IV B elements, M(NR12)2 and M(NR1R2)2 (M=Ge, Sn, or Pb; R1 =Me3Si, R2=Me3C). In: J. Chem. Soc., Chem. Commun. Nr. 21, 1974, ISSN 0022-4936, S. 895–896, doi:10.1039/C39740000895.
- ↑ Peter B. Hitchcock, Michael F. Lappert, Barry J. Samways, Erica L. Weinberg: Metal (Li, Ge II , Ge III , Sn II , and Pb II ) 2,6-dialkylbenzenethiolates; X-ray crystal structures of Sn(SAr) 2 (Ar = C 6 H 2 Bu t 3 -2,4,6) and [M(SAr′) 2 ] 3 (M = Sn or Pb, Ar′= C 6 H 3 Pr i 2 -2,6). In: J. Chem. Soc., Chem. Commun. Nr. 24, 1983, ISSN 0022-4936, S. 1492–1494, doi:10.1039/C39830001492.
- ↑ a b c Naokazu Kano, Kazusato Shibata, Norihiro Tokitoh, Renji Okazaki: Synthesis, Structure, and Reactivity of Kinetically Stabilized Divalent Organolead Compounds (Plumbylenes). In: Organometallics. Band 18, Nr. 16, August 1999, ISSN 0276-7333, S. 2999–3007, doi:10.1021/om990188z.
- ↑ a b Karl Wilhelm Klinkhammer, Wolfgang Schwarz: Bis(hypersilyl)tin and Bis(hypersilyl)lead, Two Electron-Rich Carbene Homologs. In: Angewandte Chemie International Edition in English. Band 34, Nr. 12, 7. Juli 1995, ISSN 0570-0833, S. 1334–1336, doi:10.1002/anie.199513341.
- ↑ Jonathan P. H. Charmant, Mairi F. Haddow, F. Ekkehardt Hahn, Dennis Heitmann, Roland Fröhlich: Syntheses and molecular structures of some saturated N-heterocyclic plumbylenes. In: Dalton Transactions. Nr. 43, 2008, ISSN 1477-9226, S. 6055, doi:10.1039/b808717b.
- ↑ a b c d Roland C. Fischer, Philip P. Power: π-Bonding and the Lone Pair Effect in Multiple Bonds Involving Heavier Main Group Elements: Developments in the New Millennium. In: Chemical Reviews. Band 110, Nr. 7, 14. Juli 2010, ISSN 0009-2665, S. 3877–3923, doi:10.1021/cr100133q.
- ↑ Ming-Der Su: Theoretical Study on the Reactivities of Stannylene and Plumbylene and the Origin of their Activation Barriers. In: Chemistry – A European Journal. Band 10, Nr. 23, 3. Dezember 2004, ISSN 0947-6539, S. 6073–6084, doi:10.1002/chem.200400413.
- ↑ a b Marian Olaru, Daniel Duvinage, Enno Lork, Stefan Mebs, Jens Beckmann: Heavy Carbene Analogues: Donor-Free Bismuthenium and Stibenium Ions. In: Angewandte Chemie International Edition. Band 57, Nr. 32, 6. August 2018, S. 10080–10084, doi:10.1002/anie.201803160.
- ↑ a b Norihiro Tokitoh, Wataru Ando: Silylenes (and Germylenes, Stannylenes, Plumbylenes). In: Reactive Intermediate Chemistry. John Wiley & Sons, Inc., Hoboken, NJ, USA 2005, ISBN 978-0-471-72149-9, S. 651–715, doi:10.1002/0471721492.ch14.
- ↑ Manfred Weidenbruch: From a Cyclotrisilane to a Cyclotriplumbane: Low Coordination and Multiple Bonding in Group 14 Chemistry. In: Organometallics. Band 22, Nr. 22, Oktober 2003, ISSN 0276-7333, S. 4348–4360, doi:10.1021/om034085z.
- ↑ a b Jacques Barrau, Ghassoub Rima, Tajani El-Amraoui: Stable divalent heteroleptic species ArO(X)M [Ar=2,4,6-Tris(dimethylaminomethyl)phenyl-, M=Ge, Sn, Pb]. In: Journal of Organometallic Chemistry. Band 561, Nr. 1-2, Juni 1998, S. 167–174, doi:10.1016/S0022-328X(98)00552-X.
- ↑ F. Ekkehardt Hahn, Dennis Heitmann, Tania Pape: Synthesis and Characterization of Stable N-Heterocyclic Plumbylenes. In: European Journal of Inorganic Chemistry. Band 2008, Nr. 7, März 2008, S. 1039–1041, doi:10.1002/ejic.200701260.
- ↑ Shenglai Yao, Stefan Block, Markus Brym, Matthias Driess: A new type of heteroleptic complex of divalent lead and synthesis of the P-plumbyleniophosphasilene, R2Si=P–Pb(L): (L = β-diketiminate). In: Chemical Communications. Nr. 37, 2007, ISSN 1359-7345, S. 3844, doi:10.1039/b710888e.
- ↑ Sally Brooker, Jan Karel Buijink, Frank T. Edelmann: Synthesis, structure, and reactivity of the first stable diaryllead(II) compound. In: Organometallics. Band 10, Nr. 1, Januar 1991, ISSN 0276-7333, S. 25–26, doi:10.1021/om00047a014.
- ↑ Keith Izod, Corinne Wills, William Clegg, Ross W. Harrington: Acyclic Dialkylstannylene and -Plumbylene Compounds That Are Monomeric in the Solid State. In: Organometallics. Band 28, Nr. 19, 12. Oktober 2009, ISSN 0276-7333, S. 5661–5668, doi:10.1021/om900614q.
- ↑ a b Trevor Janes, Pavel Zatsepin, Datong Song: Reactivity of heavy carbene analogues towards oxidants: a redox active ligand-enabled isolation of a paramagnetic stannylene. In: Chemical Communications. Band 53, Nr. 21, 2017, ISSN 1359-7345, S. 3090–3093, doi:10.1039/C7CC00837F.
- ↑ Julia Schneider, Kilian M. Krebs, Sarah Freitag, Klaus Eichele, Hartmut Schubert: Intramolecular Tetrylene Lewis Adducts: Synthesis and Reactivity. In: Chemistry - A European Journal. Band 22, Nr. 28, 4. Juli 2016, S. 9812–9826, doi:10.1002/chem.201601224.
- ↑ Brian P. Johnson, Stefan Almstätter, Fabian Dielmann, Michael Bodensteiner, Manfred Scheer: Synthesis and Reactivity of Low-Valent Group 14 Element Compounds. In: Zeitschrift für anorganische und allgemeine Chemie. Band 636, Nr. 7, 28. Mai 2010, S. 1275–1285, doi:10.1002/zaac.201000029.
- ↑ Martin Stürmann, Wolfgang Saak, Heinrich Marsmann, Manfred Weidenbruch: Tetrakis(2,4,6-triisopropylphenyl)diplumbene: A Molecule with a Lead–Lead Double Bond. In: Angewandte Chemie International Edition. Band 38, Nr. 1-2, 1999, ISSN 1521-3773, S. 187–189, doi:10.1002/(SICI)1521-3773(19990115)38:1/2<187::AID-ANIE187>3.0.CO;2-2.
- ↑ a b c d Shirley Hino, Marilyn Olmstead, Andrew D. Phillips, Robert J. Wright, Philip P. Power: Terphenyl Ligand Stabilized Lead(II) Derivatives: Steric Effects and Lead−Lead Bonding in Diplumbenes. In: Inorganic Chemistry. Band 43, Nr. 23, November 2004, ISSN 0020-1669, S. 7346–7352, doi:10.1021/ic049174y.
- ↑ a b Jeremy D. Erickson, James C. Fettinger, Philip P. Power: Reaction of a Germylene, Stannylene, or Plumbylene with Trimethylaluminum and Trimethylgallium: Insertion into Al–C or Ga–C Bonds, a Reversible Metal–Carbon Insertion Equilibrium, and a New Route to Diplumbenes. In: Inorganic Chemistry. Band 54, Nr. 4, 16. Februar 2015, ISSN 0020-1669, S. 1940–1948, doi:10.1021/ic502824w.
- ↑ Karl Heinz Dötz: Carbene Complexes in Organic Synthesis [New Synthetic Methods (47)]. In: Angewandte Chemie International Edition in English. Band 23, Nr. 8, August 1984, S. 587–608, doi:10.1002/anie.198405871.
- ↑ Martin Stürmann, Manfred Weidenbruch, Karl W. Klinkhammer, Falk Lissner, Heinrich Marsmann: New Plumbylenes and a Plumbylene Dimer with a Short Lead−Lead Separation. In: Organometallics. Band 17, Nr. 20, September 1998, ISSN 0276-7333, S. 4425–4428, doi:10.1021/om9804475.
- ↑ Goran Bačić, David Zanders, Bert Mallick, Anjana Devi, Seán T. Barry: Designing Stability into Thermally Reactive Plumbylenes. In: Inorganic Chemistry. Band 57, Nr. 14, 16. Juli 2018, ISSN 0020-1669, S. 8218–8226, doi:10.1021/acs.inorgchem.8b00719.
- ↑ Brian D. Rekken, Thomas M. Brown, Marilyn M. Olmstead, James C. Fettinger, Philip P. Power: Stable Plumbylene Dichalcogenolate Monomers with Large Differences in Their Interligand Angles and the Synthesis and Characterization of a Monothiolato Pb(II) Bromide and Lithium Trithiolato Plumbate. In: Inorganic Chemistry. Band 52, Nr. 6, 18. März 2013, ISSN 0020-1669, S. 3054–3062, doi:10.1021/ic302513c.